
Guidance for Non-commercial Clinical Trials
Being a sponsor in a clinical trial entails responsibilities and obligations as defined in ICH-GCP E6(R3). As a sponsor, it is essential to have an overview of the regulatory requirements as well as the processes and workflows associated with these requirements.
Look at the interactive map to get an overview of where which rules apply.
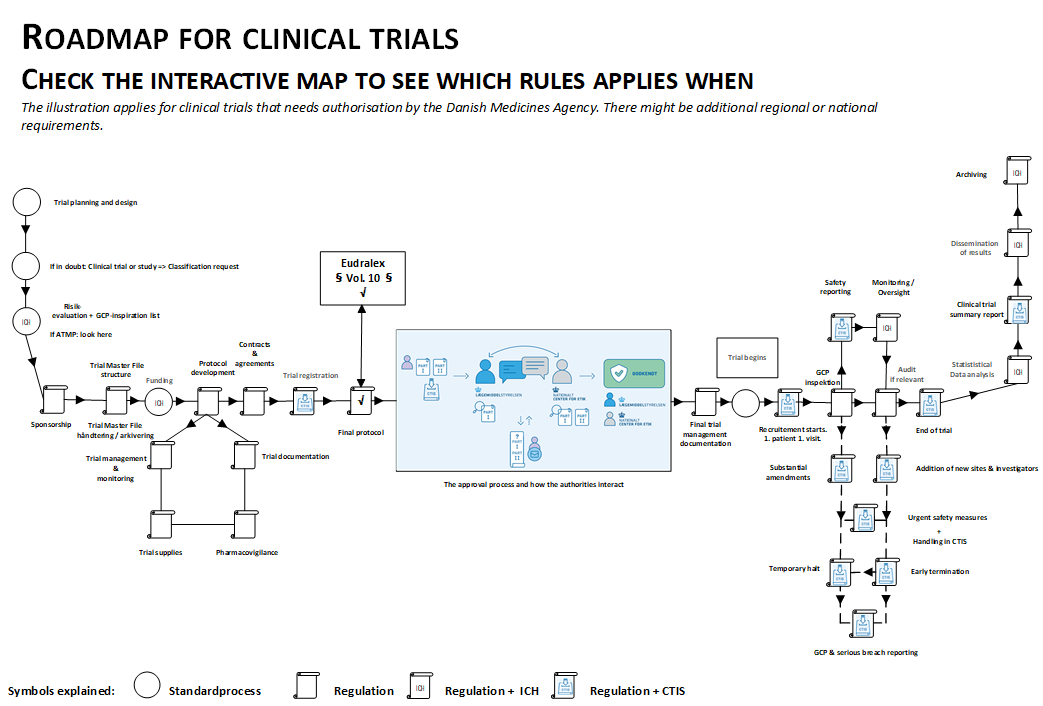
With the implementation of Regulation 536/2014 for clinical trials, authorities in Europe have been provided with working conditions that ensure a harmonized approval process, while also adhering to the established deadlines. To meet these deadlines and requirements, sponsors must be thoroughly prepared when submitting the clinical trial and ensure that quality is integrated into both the design and workflows.
As an authority, it is our experience that it can be challenging for non-commercial sponsors, who may not necessarily have regulatory experience, to gain a clear understanding of the authorities’ requirements and expectations and thus comply with them.
This page has been created to highlight the topics that may be particularly relevant or challenging for non-commercial sponsors. We also aim to point out where help can be found. As a sponsor, you are always welcome to contact the clinical trials section for further information or guidance.
Definitions On Clinical Trials
Low-intervention trials and pragmatic elements
Regulation of Low-Intervention Trials
There are no advantages for sponsors in a low-intervention trial compared to a regular clinical trial. We observe the perception that there is possibility of omitting individual medication accounting and that risk-adapted adverse event management is associated with low-intervention trials, but this is not the case.
Both of these options are available in regular clinical trials if the risk assessment and justification are described in the protocol. The evaluation time is also not different for low-intervention trials.
A Low Intervention Trial (LIT) is a regulatory term and is not the same as considering the trial as low-intervention in the clinic.
For the preparation of the protocol, we refer to “Risk Proportionate Approaches in Clinical Trials” and the Danish Medicines Agency’s guidance on risk-based adverse event management.
Criteria for Low-Intervention Trials
As a starting point, the definition of a low-intervention trial includes that the investigational medicinal product is marketed, used in accordance with the summary of product characteristics (SmPC), and does not impose more than minimal burden on the trial subjects.
The following are considered in the trial-specific justification of a low-intervention trial:
- The justification is not a clinician’s “ordinary” risk assessment, but the sponsor’s justification, which must be based on all the points in the legislation (see below).
- References to other trials classified as low-intervention cannot be used as the assessment for LIT status is trial-specific.
- It is encouraged to provide a solid justification when submitting the application. Unless the treatment guidelines in the summary of product characteristics (SmPC) are followed completely or there is a specific clinical guideline (national or European) to refer to, the sponsor is encouraged to provide a detailed justification for LIT status, based on a literature review for solid evidence, the department’s experience (including data on both the number of treated patients and the number of years), as well as a thorough safety assessment of the department’s experience and a conclusion that the evidence is in line with clinical guidelines.
- A LIT status is assessed both “clinically” and ethically, so aspects that may add extra inconvenience for the trial participant, such as additional blood samples/biopsies, etc., must be justified from both a clinical and ethical perspective.
Definition of Low-Intervention Trials in the Clinical Trials Regulation:
“A clinical low-intervention trial” means a clinical trial that fulfills all of the following conditions:
a) The investigational medicinal products, excluding placebos, are authorized.
b) According to the protocol of the clinical trial:
i) The investigational medicinal products are used in accordance with the terms of the marketing authorization, or
ii) The use of the investigational medicinal products is evidence-based and supported by published scientific evidence on the safety and efficacy of those investigational medicinal products in one of the concerned Member States.
c) The additional diagnostic or monitoring procedures pose only minimal additional risk or burden to the safety of the trial subjects compared to normal clinical practice in any concerned Member State.
What is a Pragmatic Trial?
There is no definition of a pragmatic trial in EU legislation. ICH GCP E6 (R3) describes that trials can have pragmatic elements: “Pragmatic elements in clinical trials are those that integrate aspects of clinical practice into the design and conduct of the trial (e.g., simplified protocols with streamlined data collection).”
Calling a trial pragmatic does not provide any specific regulatory advantages.
For the preparation of the protocol, we refer to “Risk Proportionate Approaches in Clinical Trials” and the Danish Medicines Agency’s guidance on risk-based adverse event management.
Sponsors Role and Responsibilities in Clinical Trials
A clinical trial must have a sponsor who assumes overall responsibility for the trial.
Each trial must clearly identify who has taken on this sponsor responsibility.
This is particularly important when a hospital or hospital department is listed as the sponsor of a clinical trial. Regulation (EU) No 536/2014 on clinical trials on medicinal products, the Danish Act on Clinical Trials, and the ICH Guideline for Good Clinical Practice (GCP) outline these obligations.
Please refer to the Q&A document (only in Danish), which describes the Danish Medicines Agency’s expectations regarding the sponsor’s role in terms of organisation and responsibility.
Additional definition information on clinical trials with medicinal products
Templates for the Application
Contract Templates
Protocol Template (October 2025)
We have developed a new protocol template that includes even more guidance from both the Scientific Ethical Medical Committees and the Danish Medicines Agency.
By using this new template, we ensure that the regulatory requirements for the protocol are more thoroughly met – and hopefully, make the protocol development both easier and better.
We encourage you to use this protocol template (instead of a previously used template in the department).
Technical guidance (CTIS a.o.)
General Information
Below are the errors and/or deficiencies most commonly observed during the validation of a trial application or substantial modifications, which consequently trigger considerations for the sponsor.
With this list, we hope to prevent errors and facilitate the process.
We would also like to point out that the sponsor is not charged a fee until the submission is declared valid. Therefore, no fee is required if the submission is rejected by the authorities or if the sponsor's deadlines expire during this phase.
Please find more information on our general Q&A for clinical trials.
Action-points from the GCP-inspectors
Over a period of time, the Danish Medicines Agency's GCP inspectors have conducted a project focusing on the management of systems and data in investigator-initiated trials. The project has covered many areas, some of the most important being tasks contracted out, such as the provision of systems for randomization and data collection, as well as documentation of processes after the last trial participant has left the trial, including data analysis, unblinding, and documentation of activities in the Trial Master File.
The project was carried out both to increase researchers' and regions' focus on obtaining valid conclusions from the trials and to provide guidance and set standards in challenging areas that have not been frequently inspected before.
Structured Data
1. Update of existing documents to new versions:
To upload a new version of a document, use the ”Update” button (Paper-ikon) . Do not use the ”Add document”, button, which is only intended for new documents.
2. Versioning and naming of documents:
Correct naming of documents and date/versioning are entered in the structured data fields in CTIS. Avoid entering the date/version in the very file name.
Please also see the CTCG best practice guide on naming of documents on the HMA homepage under key documents.
Section Form
1. Template for the cover letter:
We recommend to use the Cover Letter Template (From CTCG); Initial application, Substantial Modification, Modification description
The latest version can be found on CTCG’s homepage under "Key documents list".
2. Invoice Information:
Clearly state the hospital department, EAN and CVR number, + special references considered important on the invoice, if any.
Section Part I
Section Part I, Trial details, Protocol information
1. Indication of names in the protocol:
Only the sponsor's name should be stated in the protocol. Names of co-investigators, collaborators, laboratories, etc. should be omitted from the protocol to avoid unnecessary substantial modifications if these change.
Sectionen Part I, Sponsors
2. Monitor:
Remember to indicate monitor in Third parties. See section 2.6 in the GCP units guidance.Sectionen Part I, Products
3. The field "Excluded MSCs":
This field is only used for multinational trials, where not all medicinal products are used in all countries.4. Upload of the product resumé (SmPC)
For trials using only marketed medicinal products within the indication, it is only necessary to submit their respective Summary of Product Characteristics (SmPC), which should only be uploaded in its associated tab under the Investigator's Brochure. It is not necessary to upload it more than once, or under other tabs, even though these tabs may be marked with a star symbol.
Non-substatial modifications and how they are uploaded in CTIS
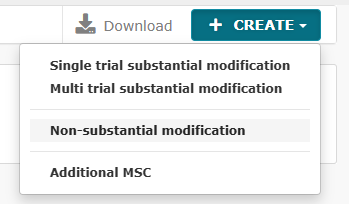
It is now possible to submit non-substantial modifications, where it earlier required submission of a substantial modification.
It includes, amongst others, the extension of the clinical trial duration.
See the full list of new functionalities further down on this page.
There are two types of non-substantial modifications:
- Non-substantial modifications (NSM)
- 81.9 non-substantial modifications (81.9 NSM)
81.9 NSMs must be continuously updated by the sponsor in CTIS throughout the trial period.
Other NSMs must be updated in CTIS together with the next substantial modification (SM), or at the latest at the end of the trial if no SMs have been submitted in the meantime.
81.9 NSMs are changes to a clinical trial that are not substantial modifications (SMs), but which are nevertheless relevant for the supervising activities of the concerned Member States. These must be continuously updated by the sponsor in the EU database in accordance with Article 81(9) of Regulation (EU) No 536/2014. For a list of non-substantial modifications, see Annex IV of the document CLINICAL TRIALS REGULATION (EU) NO 536/2014, VERSION 7.1.
It is always the sponsor’s responsibility to assess whether a change is non-substantial (i.e., that it does not affect the scientific value of the trial or the rights and safety of the participants). It is important that this assessment can be justified during monitoring visits and any inspections.
All non-substantial modifications, including both 81.9 NSMs and NSMs, do not require assessment or approval by the authorities before implementation.
See the full list of CTIS functionalities for non-substantial modifications below:
Tab MSCs:
- Trial subject numbers
Tab Part I:
- Trial information - following sections can be edited;
- Medical condition
- Trial duration
- Source of monetary support or material support
- Age range (minors cannot be added)
- Clinical trial group (Healthy, patients, vulnerable)
- Protocol
- All sections can be edited, new documents can be uploadet, existing documents can be opdated
- All sections can be edited, new documents can be uploadet, existing documents can be opdated
- SA and PIP
- SA can be added/updated
- PIP can be updated, if there is an existing PIP, but no new PIP can be added
- Associated CTs
- Associated CTs can be added/deleted
- Document agreement from another sponsor, can be added/updated
- Sponsor
- Sponsor informations can be changed, as long as it in not ORG-ID
- Co-sponsor can be deleted, but not added
- Responsibilities can be changed
- Contact points can be added/changed
Safety reports
Annual safety reports
Annual Safety Report (ASR)
The annual safety report must be submitted via CTIS. Please note that the correct user role is required for the Annual Safety Reporting tab to be visible. See our guide if needed.
For mono-national trials conducted only in Denmark, the GCP unit's template for annual safety reports should be used, which can be found here: Incidents and Adverse Reactions Guidelines and Templates - GCP Units.
For multinational trials (sites both in Denmark and abroad), the ASR template from the Heads of Medicines Agency (HMA, CTCG) should be used, which can be found here: Heads of Medicines Agencies: Key Document List => Clinical Trials Coordination Group under Clinical Trial Safety, Simplified Template of Annual Safety Report.
SUSARS
SUSARs
According to the new EU regulation, SUSARs must be reported directly to the European EudraVigilance database. Read more here.
Other safety reports
Other Safety Reports
If there is a need to notify the Danish Medicines Agency about other safety issues such as unexpected events, urgent safety measures, and serious breaches, the Notifications tab should be used.
See CTIS screenshot.
Temporary Halt and Early Termination (pEOT)
Temporary halts and early termination of trials due to safety reasons must be recorded in the Notifications tab.
See CTIS screenshot.
Change Log
February 9, 2026: Information on submission of non-substantial modifications in CTIS
February 4, 2026: Information on 81.9 non-substantial modifications in CTIS
November 4, 2025: Q&A on Sponsors responsibility
October 2025: New protocol template reflecting revised ICH GCP standard.
July 11, 2025: Roadmap links to latest CTIS guidance.