
Development of medical devices
Medical devices are products which are used to diagnose, prevent relieve or treat a disease, disability, injury, etc. There are more than 500,000 different types of medical devices available, covering anything from wheelchairs and glasses to pacemakers, mobile phone apps and state-of-the-art surgical equipment.
Read about the phases that medical devices must go through before and after they are placed on the market. Click the arrows to expand the text.
Idea generation
An idea for a new medical device or an in vitro diagnostic medical device begins to form. The idea could come from a doctor, a nurse, a researcher, a medical device company or a member of the public who has come up with something clever.
The first step is to determine if the idea can be implemented and will typically involve close collaboration with engineers and other experts. If the idea can be implemented, the device must then be prototyped – usually in collaboration with a medical device company that will finance the development of the medical device.
Test phase
If a new medical device is to be used for patients, it must work as intended and be safe to use. From the very start, the manufacturer must perform tests to document the performance and safety of the device and show that the benefits of using it outweigh any potential risks. For the sake of patient safety, implanted devices and devices in risk class III (MDR) must be subjected to clinical investigation, including where appropriate testing in humans. For medical devices in other risk classes, it may be necessary to conduct a clinical investigation in humans if there are not enough clinical data to provide evidence of the device’s performance and safety. In the case of in vitro diagnostic medical devices, it may be necessary to conduct a clinical investigation of the device’s performance to provide evidence of the performance and safety of the device.
These investigations are included in a risk analysis and a clinical evaluation (MDR) or a performance evaluation (IVDR), which are key elements of the manufacturer's technical documentation of the device’s performance and safety. Besides the manufacturer's own tests and clinical data, the documentation can also include knowledge from scientific articles. The documentation of the device must be updated regularly.
If the manufacturer is able to demonstrate to the notified body that the medical device is equivalent to an existing device, and the manufacturer can also provide evidence of the safety and performance of the equivalent device, the manufacturer does not have to perform all investigations again before placing the product on the market. This is called equivalence.
Clinical evaluation of medical devices, etc.
Before a device can be placed on the market and used in the EU/EEA, the manufacturer must perform a clinical evaluation. This applies to all devices regardless of risk class. The requirements for clinical evaluation are described in article 10 and article 61 of Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices. Further guidance is also provided in the MDCG guidelines on the website of the European Commission "MDCG 2020-5 Guidance on clinical evaluation – Equivalence” and ”MDCG 2020-6 Guidance on sufficient clinical evidence for legacy devices”.
Clinical evaluation is a process related to the manufacturer’s quality management. In this process, the manufacturer plans and collects clinical data from humans from various sources, e.g. from scientific journals and clinical investigations, and subsequently analyses and evaluates these data. The clinical evaluation must show that the risks associated with using the device are acceptable relative to the performance of the device. The evaluation must also ensure that there is sufficient clinical evidence to demonstrate that the medical device is safe and has the performance and benefits that the manufacturer claims it to have.
In the clinical evaluation, the manufacturer reviews and assesses clinical data, e.g. data from clinical investigations that have tested the device in humans, relevant literature and articles and the like from scientific journals, clinically relevant information from market surveillance, empirical data from corresponding products and, where relevant, information from the manufacturer’s post-market clinical follow up (PMCF), see also the PMCF section below. The information must be reviewed, analysed and evaluated in the clinical evaluation so that the manufacturer can demonstrate that the device meets the safety and performance requirements.
The documentation of the clinical evaluation must be included in the combined technical documentation which the manufacturer must be able to present.
Equivalence
If the manufacturer uses clinical data from, for example, scientific literature whose data relate to a device other than the device subjected to the clinical evaluation, the manufacturer must be able to demonstrate in the clinical evaluation that the device is equivalent to (i.e. corresponds to) the device to which the data relate.
The manufacturer must be able to demonstrate that there is no material difference between the two devices with respect to device safety and performance; nor with respect to the technical, biological and clinical properties. The manufacturer must also be able to provide evidence that the level of data has been sufficient to demonstrate that the two devices are equivalent.
Consideration of device equivalence is a separate process of the clinical evaluation. It is conducted to determine if and the degree to which the clinical data of the equivalent device can be used in the clinical evaluation of the device in question.
If the clinical data available in literature, for example, are insufficient to document the safety and performance of the device in question, it may be necessary to conduct a clinical investigation to obtain clinical data for the device.
Implants and class III devices
If clinical data from a clinical investigation of an equivalent device are available, the manufacturer of implantable devices and class III devices (e.g. breast implants or pacemakers) does not have to perform a new clinical investigation. This requires that the two manufacturers (of the new device and the equivalent device, respectively) enter into a contract that will ensure that the manufacturer of the new device has ongoing access to the technical documentation of the equivalent device to which the clinical data relate. In addition, the original clinical evaluation must have been performed in compliance with the requirements of the regulation.
Data from clinical investigations
The using data from clinical investigations as evidence of safety and performance, a critical evaluation of the results of all available clinical investigations of relevance to the device must be performed.
In the case of implantable devices and class III devices (e.g. breast implants and pacemakers), a clinical investigation must generally always be performed unless the already existing clinical data are sufficient to demonstrate that the device meets the general safety and performance requirements, see the section on equivalence for implants and class III devices.
As part of the clinical evaluation, the manufacturer must critically review and assess the clinical data and assess which data are suitable to establish the safety and performance of the device. Based on an assessment of the relevant clinical data, the manufacturer must conclude if the device meets the general safety and performance requirements.
Evaluation plan
To organise and document a clinical evaluation, the manufacturer must establish an evaluation plan, e.g. describing
- the manufacturer’s intended purpose of the device
- the general safety and performance requirements that require support from clinical data
- the intended target group and
- the methods used to determine if the benefit-risk ratio is acceptable.
The evaluation plan must include a clinical development plan for the product. This plan is to describe how the manufacturer plans the course of clinical investigations – from the first-in-man studies to the manufacturer’s post-market clinical follow up.
The clinical evaluation must be thorough and objective and take into account both favourable and unfavourable data.
Evaluation report
After the manufacturer has reviewed and assessed all relevant clinical data, the results of the clinical evaluation and the clinical evidence must be gathered in a clinical evaluation report. The report is part of the technical documentation.
The report must include a conclusion on the performance of the device, including if the device has the performance claimed by the manufacturer. The conclusion must also describe the safety of the device and whether the risk of any side effects and unintended follow-on effects of using the device is acceptable relative to the intended performance of the device.
The manufacturer must continuously update the report throughout the lifetime of the device, and must do so as new knowledge related to the safety and performance of the device becomes available. Any new knowledge must also be included in the risk analysis.
In the case of devices that have been used for many years or variants of such devices, it may be sufficient to assess the existing evidence of clinical experience to fulfil he requirement for clinical data.
Post-market clinical follow-up (PMCF)
The PMCF process implies that the manufacturer must proactively collect and evaluate clinical data from the use of the device after the device has obtained CE marking and has been placed on the market. The PMCF process is to confirm the safety and performance of the device throughout its lifetime and to make sure that any risks associated with using the device are still acceptable.
The manufacturer must establish a plan for proactively collecting and evaluating clinical data, e.g. by keeping abreast of new clinical experience, feedback from users, screening of scientific literature, PMCF register/literature studies or PMCF clinical investigations or other clinical data.
The results and evaluation must be analysed and documented in a PMCF evaluation report. In the case of class III devices, the PMCF evaluation report must be updated annually.
The clinical evaluation, including the PMCF process, the risk management processes and the post-market surveillance process are inter-dependent and must be updated regularly.
Development and risk classification
Early in the development phase, the manufacturer must determine which risk class the device belongs to. The risk classes indicate, for example, which safety and performance risk requirements that must be met before the manufacturer can place the device on the Danish market.
While class I (MDR)/class A (IVDR) is associated with the lowest risk, class III/class D is associated with the highest risk. A class I medical device could be a walker, whereas breast implants are class III medical devices. By the same analogy, a sample container is a class A device, and a HIV test is a class D device.
The risk class is determined based on the device’s intended purpose. It depends on factors such as for how long and how often the device is to be used, if it is a device to be implanted in the body, or if, for example, it is a self-testing device. See the triangle with the medical device risk classes.
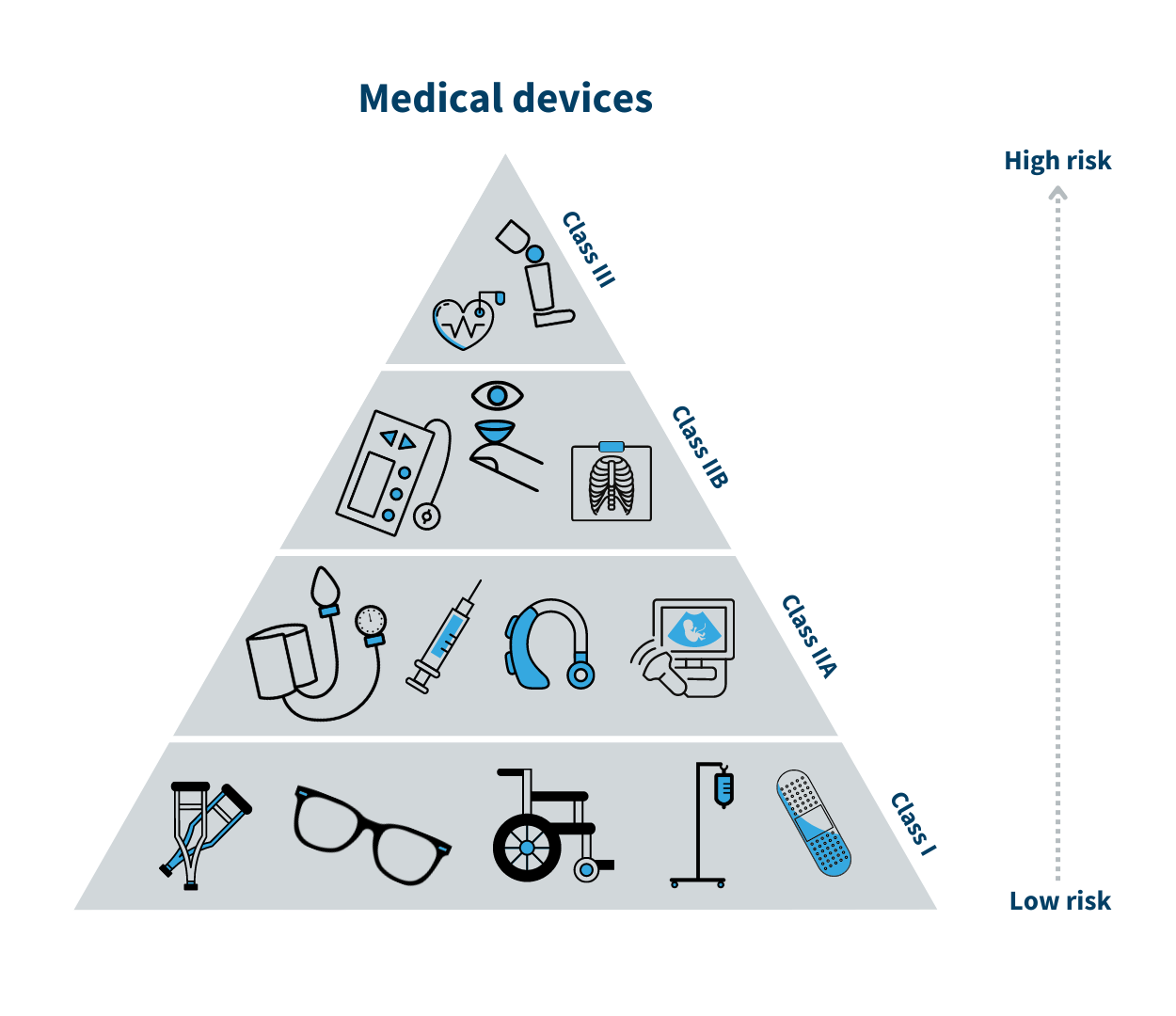
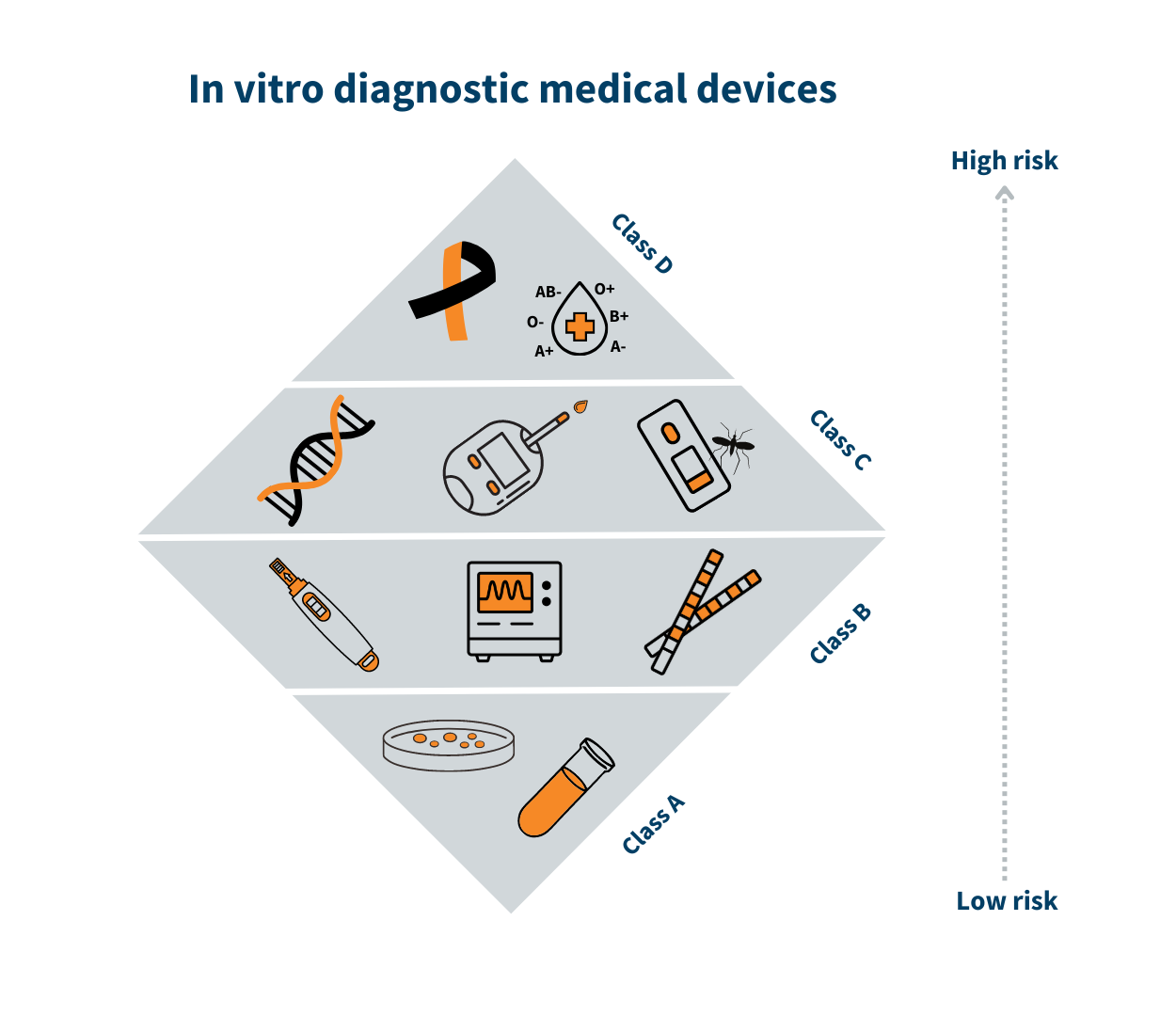
If the device developed by the manufacturer belongs to class I/class A, it is the manufacturer who is responsible for monitoring the efficacy and safety of the device. If the device belongs to the medium- or high-risk classes (class IIa, IIb and III/classes B, C and D), the technical documentation of the device must be reviewed and assessed by external experts of a so-called notified body, which is a private organisation authorised by the Danish Medicines Agency.
Marketing and safety monitoring of medical devices
Medical devices must be CE marked before they can be placed on the market. CE marking shows that the device complies with EU legislation. If a medical device is in a higher risk class than class I/class A, then a so-called notified body must assess if the device can be CE marked. The notified body assesses if the documentation for the product's safety and performance is sufficient for the product to be CE marked. Once a certificate, if any, has been issued and the manufacturer has signed a declaration of conformity, the manufacturer may affix the CE marking to its device and place the product on the market.
When the device has been placed on the market, the manufacturer must continue to monitor the device. This implies having a system for assessing complaints and reporting serious incidents to relevant competent authorities and documenting any such events.
Healthcare professionals, manufacturers, authorised representatives, distributors and importers are obligated to report serious incidents (defects, failure or deficiencies) with medical devices. Manufacturers, healthcare professionals and individuals with operational responsibility for public and private hospitals must report the incidents to the competent authority directly. Whereas authorised representatives must report incidents to the manufacturer, importers and distributors must report incidents to the manufacturer or possibly the importer or the authorised representative.
The general public can also report serious incidents and suspected serious incidents to the Danish Medicines Agency.
The Danish Medicines Agency reviews all reports to decide with the manufacturer if the manufacturer should make changes to the device, update the instructions for use or if, in the last resort, the device should be recalled from the market. We collaborate in this matter with the authorities of other countries, including those in the EU.
Ongoing recertification of medical devices in medium- and high-risk classes
Medical devices in medium- and high-risk classes (IIa, IIb and III/classes B, C and D) must be recertified regularly. This means that the documentation of the device must be reviewed by experts in notified bodies at regular intervals. The review of the documentation must take place at least every five years to ensure the device is still safe to use and works as intended.