
Notification duty
The notification duty established in the articles of the regulation depend on whether or not the device is CE marked and whether the intention is to use the collected data for conformity assessment of the device.
The MDR sets out three different types of clinical investigations of medical devices:
Article 62 “Product development”: Investigation of clinical devices to assess the safety and performance of a device, either for a non-CE marked medical device or a CE marked medical device for which the manufacturer intends to expand the product’s intended purpose.
Article 74(1) “PMCF investigation” involving additional invasive/burdensome procedures”: Manufacturer-initiated post-market follow-up studies of CE marked devices in which the subjects are submitted to additional invasive/burdensome procedures.
Article 82 “Other investigations”
The figure below is from the European Commission’s guidance document “MDCG 2021-6 Rev. 1 Regulation (EU) 2017/745 – Questions & Answers regarding clinical investigation”. The guidance document is available for download here.
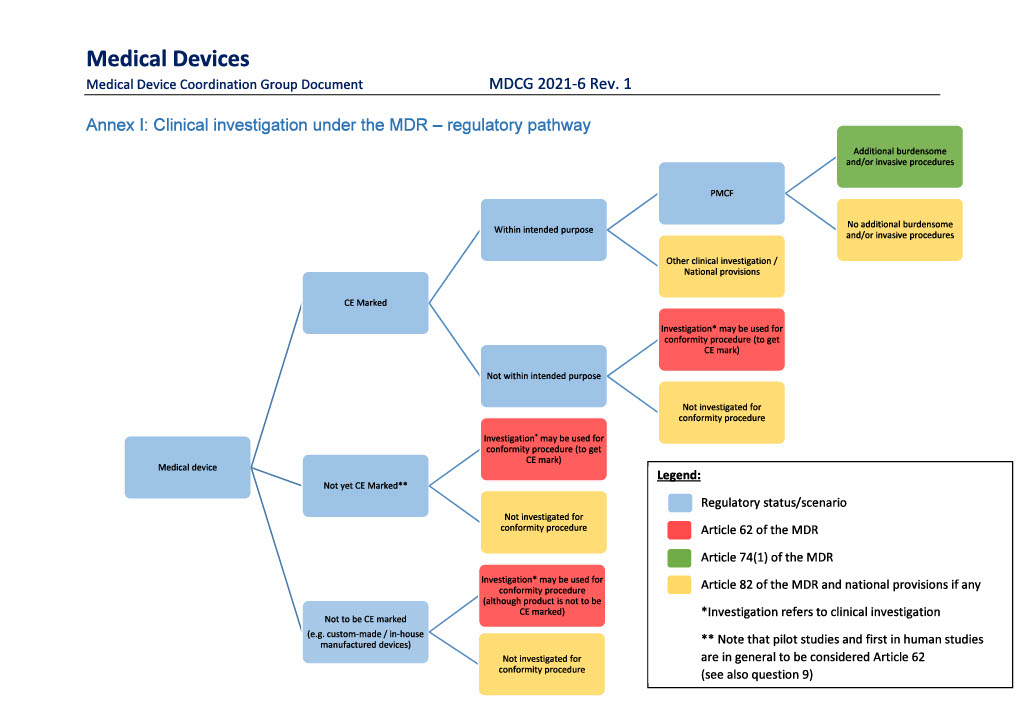

Article 62 – Product development
It is established for CE marked medical devices that if the clinical investigation is carried out as part of the clinical evaluation of the device for subsequent purposes of conformity assessment (process to demonstrate whether the requirements of the regulation relating to a device have been fulfilled), and one or more of the following purposes is
- to establish and verify that a device is designed in such a way that it is suitable for one or more of the device’s specific purposes and achieves the performance intended;
- to establish and verify the clinical benefits; or
- to establish and verify the clinical safety of the device and to determine any undesirable side effects and assess whether they constitute acceptable risks when weighed against the benefits to be achieved by the device;
the investigation must be authorised by the Danish Medicines Agency before the clinical investigation may commence.
Specific national rules have been introduced in Denmark, which mean that clinical investigation must be authorised regardless of the class of the device.
Clinical investigation in which a CE marked device is used outside the scope of its intended purpose and in which the investigation is carried out with the intention of expanding or modifying the device’s intended purpose must be notified under article 62.
An application for authorisation of clinical investigation under article 62 must be submitted simultaneously to the Danish Medicines Agency and the Medical Research Ethics Committees as one single application. The common application form is available for download on this page.
Article 74(1) – PMCF investigation with additional invasive or burdensome procedures
Clinical investigation of CE marked devices in which the medical device is used within the scope of its intended purpose, and the clinical investigation is part of the manufacturer’s post-market clinical follow-up (PMCF) obligations, and where the investigation implies submitting the subjects to additional burdensome/invasive procedures, is subject to assessment by the Medical Research Ethics Committees under article 74(1). The investigation must be notified to the Danish Medicines Agency 30 days before its commencement. Find out more about how sponsors may notify the Danish Medicines Agency here.
Clinical investigation of CE marked devices within the scope of its intended purpose as part of the manufacturer’s PMCF plan could for example be investigations with the purpose of collecting clinical data to study the effect or side effects when the device is used over a longer period of time, and where it might include additional burdensome/invasive procedures (such as a biopsy or the like) that normally would not be carried out under normal clinical use of the device.
Whether additional procedures are burdensome for the subjects must be determined from the perspective of the subjects. Additional procedures which are burdensome could be procedures which may cause an increased risk of complications or side effects, see MDCG 2021-6 Regulation (EU) 2017/745 – Questions & Answers regarding clinical investigation.
An application for authorisation of clinical investigation under article 74(1) must be submitted to the Medical Research Ethics Committees, and the Danish Medicines Agency must be notified of the investigation 30 days before commencement of the investigation.
Article 82 - Other investigations
Clinical investigations that are not performed pursuant to any of the above purposes fall under the provisions of article 82, which establishes a number of minimum requirements for other types of clinical investigations of medical devices.
Article 82(1) specifies the following among other things: A sponsor for the investigation must be established, a positive opinion about the investigation must be issued by the Medical Research Ethics Committees, subjects must be protected and must consent to participation, and the manufacturer of the device must document that the device meets the general safety and performance requirements set out in Annex I of the regulation apart from the aspects covered by the clinical investigation.
An application for authorisation of clinical investigation under article 82 must be submitted to the Medical Research Ethics Committees.