
Udvikling af medicinsk udstyr
Medicinsk udstyr er produkter, der bruges til at diagnosticere, forebygge, lindre eller behandle sygdomme, handicap eller skader. Der findes mere end 500.000 forskellige typer medicinsk udstyr, som kan være alt fra kørestole og briller til pacemakere, apps på mobiltelefoner og avanceret operationsudstyr.
Læs om de faser, som medicinsk udstyr skal gennemgå, før og efter det er kommet på markedet. Klik på pilene og fold teksten ud.
Idéfase
En idé til et nyt medicinsk udstyr eller medicinsk udstyr til in vitro-diagnostik opstår. Ideen kan komme fra en læge, en sygeplejerske, en forsker, en medicovirksomhed eller en borger, der har fået en god idé.
Første step er at finde ud af, om ideen overhovedet kan realiseres. Det vil typisk betyde samarbejde med ingeniører og andre eksperter. Hvis ideen kan gennemføres, skal der laves en prototype af udstyret – typisk i samarbejde med en medicovirksomhed, som finansierer udviklingen af det medicinske udstyr.
Testfase
Før nyt udstyr må bruges til patienter, skal det virke efter hensigten og være sikkert at bruge. Fabrikanten skal helt fra starten af udviklingsfasen lave tests, der skal dokumentere udstyrets ydeevne og sikkerhed, og at fordelene ved at bruge udstyret opvejer evt. risici. For udstyr, der skal indopereres, og udstyr i risikoklasse III (MDR), skal der for patientsikkerhedens skyld udføres kliniske afprøvninger på mennesker. For medicinsk udstyr i andre risikoklasser kan det være nødvendigt at udføre en klinisk afprøvning på mennesker, såfremt der ikke er tilstrækkelig klinisk data til at dokumentere udstyrets ydeevne og sikkerhed. For medicinsk udstyr til in vitro-diagnostik kan det være nødvendigt at udføre en undersøgelse af ydeevne for at dokumentere udstyrets ydeevne og sikkerhed.
Testene indgår i en risikoanalyse og en klinisk evaluering (MDR) eller ydeevneevaluering (IVDR), der er centrale dele af fabrikantens tekniske dokumentation for udstyrets ydeevne og sikkerhed. Dokumentationen kan ud over fabrikantens egne test og kliniske data også omfatte viden fra videnskabelige artikler. Udstyrets dokumentation skal opdateres løbende.
Hvis fabrikanten over for det bemyndigede organ kan dokumentere, at det medicinske udstyr er magen til et, der allerede findes i forvejen, og at fabrikanten også kan fremlægge dokumentation for sikkerheden og ydeevnen af det produkt, som det er magen til, behøver fabrikanter af medicinsk udstyr ikke at lave alle tests forfra, før et produkt kan komme på markedet. Dette kaldes ækvivalens.
Klinisk evaluering af medicinsk udstyr mv.
Inden udstyr kan markedsføres og ibrugtages i EU/EØS, skal fabrikanten have udført en klinisk evaluering. Det gælder alt udstyr, uanset hvilken risikoklasse udstyret er i. Kravene til en klinisk evaluering står beskrevet i artikel 10 og artikel 61 i Europa-Parlamentets og Rådets forordning (EU) 2017/745 af 5. april 2017 om medicinsk udstyr. Der er også vejledning på EU-kommissionens hjemmeside i MDCG-vejledningerne: "MDCG 2020-5 Guidance on clinical evaluation – Equivalence” og ”MDCG 2020-6 Guidance on sufficient clinical evidence for legacy devices”.
Den kliniske evaluering er en proces i forbindelse med fabrikantens kvalitetsstyring, hvor fabrikanten planlægger og indsamler kliniske data fra mennesker fra forskellige kilder, bl.a. fra videnskabelige tidsskrifter og kliniske afprøvninger, for derefter at analysere og vurdere data. Den kliniske evaluering skal vise, at de risici, der kan være forbundet med anvendelsen af udstyret, er acceptable i forhold til udstyrets ydeevne. Evalueringen skal også sikre, at der er tilstrækkelig klinisk evidens for, at det medicinske udstyr er sikkert, og at udstyret har den ydeevne og de fordele, som fabrikanten har anført for udstyret.
En klinisk evaluering foregår ved, at fabrikanten ser på og vurderer kliniske data, f.eks. data fra kliniske afprøvninger af udstyret på mennesker, relevant litteratur og artikler e.l. fra videnskabelige tidsskrifter, klinisk relevante informationer fra markedsovervågning, erfaringsdata fra tilsvarende produkter og evt. information fra fabrikantens kliniske opfølgning efter produktet er bragt i omsætning, se også nederste afsnit om PMCF. Oplysningerne skal gennemgås, analyseres og evalueres i den kliniske evaluering, så fabrikanten kan påvise, at udstyret lever op til kravene til sikkerhed og ydeevne.
Dokumentationen for den kliniske evaluering skal indgå som en del af den samlede tekniske dokumentation, som fabrikanten skal kunne fremvise.
Ækvivalens
Hvis fabrikanten anvender kliniske data fra fx videnskabelig litteratur, hvor data stammer fra et andet udstyr end det udstyr, som den kliniske evaluering omhandler, skal fabrikanten kunne påvise i den kliniske evaluering, at udstyret er ækvivalent med (dvs. svarer til) det udstyr, som dataene handler om.
Fabrikanten skal kunne vise, at der ikke er nogen væsentlig forskel på de to udstyr med hensyn til udstyrets sikkerhed og ydeevne, heller ikke hvad angår tekniske, biologiske og kliniske egenskaber. Fabrikanten skal også kunne dokumentere, at der har været tilstrækkeligt med data til stede til at kunne påvise, at de to udstyr er ækvivalente (svarer til hinanden).
At vurdere to udstyrs ækvivalens er en separat proces i den kliniske evaluering, som laves for at afgøre hvorvidt og i hvilken grad de kliniske data fra det ækvivalente udstyr kan anvendes i den kliniske evaluering af det pågældende udstyr.
Hvis der ikke er tilstrækkelig klinisk data tilgængelig fra fx litteratur, der kan dokumentere sikkerhed og ydeevne for det pågældende udstyr, kan det være nødvendigt at udføre en klinisk afprøvning for at få kliniske data for udstyret.
Implantater og klasse III-udstyr
Hvis der findes klinisk data fra en klinisk afprøvning til et ækvivalent udstyr, kan fabrikanten af implantabelt udstyr og udstyr i klasse III (fx brystimplantater eller pacemakere) undlade selv at foretage en ny klinisk afprøvning. Det kræver at der indgås en aftale imellem de to fabrikanter (fabrikanten af et nyt udstyr og fabrikanten af det ækvivalente udstyr), så det sikres, at fabrikanten af det nye udstyr løbende har adgang til den tekniske dokumentation for det ækvivalente udstyr, som de kliniske data stammer fra. Samtidig skal den originale kliniske evaluering være udført i overensstemmelse med kravene i forordningen.
Data fra kliniske afprøvninger
Ved anvendelse af data fra kliniske afprøvninger som dokumentation for sikkerhed og ydeevne, skal der foretages en kritisk evaluering af resultaterne af alle tilgængelige kliniske afprøvninger, der er relevant for udstyret.
For implantabelt udstyr og udstyr i klasse III (fx brystimplantater eller pacemakere) skal der som udgangspunkt altid udføres kliniske afprøvninger, medmindre allerede eksisterende kliniske data er tilstrækkelige til at kunne godtgøre, at udstyret lever op til de generelle krav til sikkerhed og ydeevne, se ovenstående afsnit om ækvivalens for implantater og klasse lll-udstyr.
Fabrikanten skal i forbindelse med den kliniske evaluering lave en kritisk gennemgang og vurdering af de kliniske data og vurdere hvilke data, der er egnet til at fastlægge udstyrets sikkerhed og ydeevne. Ud fra en vurdering af den relevante kliniske data skal fabrikanten konkludere, om udstyret overholder de generelle krav til sikkerhed og ydeevne.
Evalueringsplan
For at kunne planlægge og dokumentere den kliniske evaluering skal fabrikanten lave en evalueringsplan, som blandt andet beskriver
- fabrikantens erklærede formål
- hvilke generelle krav om sikkerhed og ydeevne, der kræver støtte fra kliniske data
- hvem der er målgruppen og
- hvilke metoder, der er anvendt til at fastslå om forholdet mellem eventuelle fordele og risici er acceptabelt.
Evalueringsplanen skal bl.a. indeholde en klinisk udviklingsplan for produktet. Den skal belyse, hvordan fabrikanten planlægger forløbet af kliniske afprøvninger – fra de første afprøvninger i mennesker til fabrikantens plan for klinisk opfølgning efter udstyret er bragt i omsætning.
Den kliniske evaluering skal være grundig og objektiv og tage hensyn til både positive og negative data.
Evalueringsrapport
Når fabrikanten har gennemgået og vurderet alle relevante kliniske data, skal resultaterne af den kliniske evaluering og den kliniske dokumentation samles i en klinisk evalueringsrapport. Rapporten indgår i den tekniske dokumentation.
Rapporten skal bl.a. indeholde en konklusion om udstyrets ydeevne, herunder om udstyret har den ydeevne, som fabrikanten har angivet. Konklusionen skal også beskrive udstyrets sikkerhed, og om risikoen for enhver bivirkning og uønsket følgevirkning ved brugen af udstyret står i et acceptabelt forhold til den angivne ydeevne.
Fabrikanten skal løbende opdatere rapporten gennem udstyrets levetid. Det skal gøres efterhånden som ny viden, der vedrører udstyrets sikkerhed og ydeevne, bliver tilgængelig. Denne nye viden skal også indgå i risikoanalysen.
For udstyr, der har været anvendt i en længere årrække, og for udstyr, som er varianter af den slags udstyr, kan en vurdering af eksisterende dokumenterede kliniske erfaringer være tilstrækkeligt til at dække kravet til kliniske data.
Klinisk opfølgning, efter udstyret er bragt i omsætning (PMCF)
PMCF er en engelsk forkortelse for post market clinical follow up. PMCF-processen indebærer, at fabrikanten proaktivt skal indsamle og evaluere kliniske data fra brugen af udstyret, efter at udstyret er CE-mærket og markedsført. Det gøres for at bekræfte, at udstyret overholder kravene til sikkerhed og ydeevne i hele dets levetid, og for at sikre at eventuelle risici ved at bruge udstyret fortsat er acceptable.
Fabrikanten skal udarbejde en plan for, hvordan der proaktivt indsamles og vurderes kliniske data fx ved løbende at holde sig orienteret om nye kliniske erfaringer, feedback fra brugere, gennemgang af videnskabelig litteratur, PMCF-register/litteraturstudier eller PMCF-kliniske afprøvninger eller andre kliniske data.
Resultaterne af vurderingen skal analyseres og dokumenteres løbende i en rapport (PMCF-evalueringsrapport). For udstyr i klasse III skal PMCF-evalueringsrapporten opdateres årligt.
Den kliniske evaluering, herunder PMCF-processen, risikostyringsprocesserne og markedsovervågningsprocessen efter udstyret er markedsført, er indbydes afhængige processer, som alle løbende skal opdateres.
Udvikling og risikoinddeling
Fabrikanten skal tidligt i udviklingen af det medicinske udstyr finde ud af, hvilken risikoklasse, udstyret tilhører. Risikoklasserne bruges bl.a. til at anvise hvilke krav til sikkerhed og ydeevne, der skal være opfyldt, før fabrikanten kan markedsføre udstyret i Danmark.
Klasse I (MDR)/Klasse A (IVDR) er forbundet med den laveste risiko, mens klasse III/Klasse D er forbundet med den højeste risiko. Klasse I er eksempelvis en rollator, mens brystimplantater er klasse III. Tilsvarende er klasse A eksempelvis en prøvebeholder, mens HIV-test er klasse D.
Risikoklassen fastsættes på baggrund af udstyrets erklærede formål og afhænger bl.a. af hvor længe og hyppigt udstyret skal bruges, om det er udstyr, som skal indopereres i patientens krop, og om det f.eks. er udstyr til selvtest. Se trekanten med risikoklasserne indenfor medicinsk udstyr.
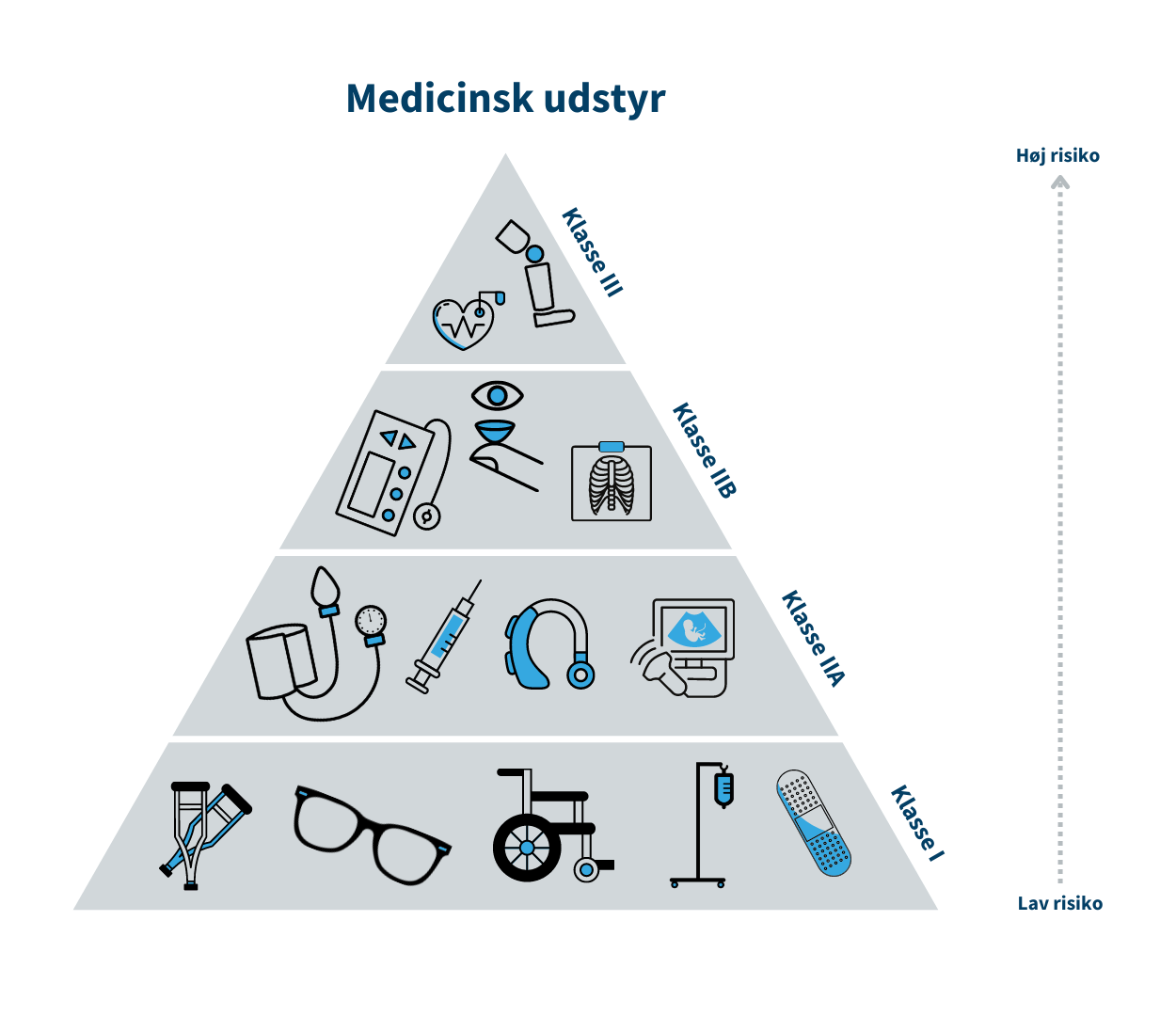
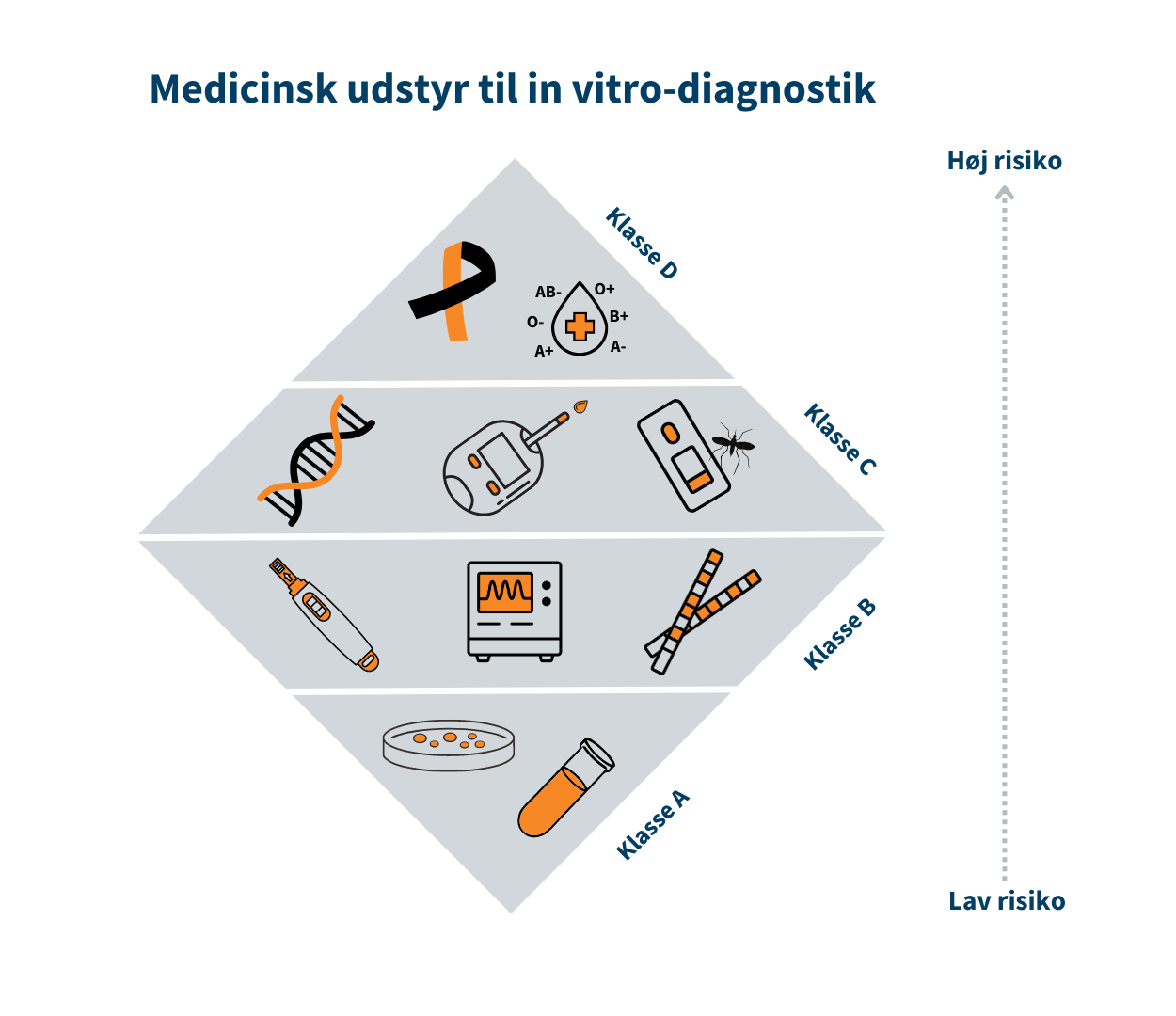
Hvis det udstyr, fabrikanten udvikler, tilhører klasse I/klasse A, skal fabrikanten selv kontrollere udstyrets effekt og sikkerhed. Hvis udstyret tilhører mellem- eller højrisikoklassen (klasse IIa, IIb og III/ klasse B, C og D), skal udstyrets tekniske dokumentation gennemgås og vurderes af eksterne eksperter i et såkaldt bemyndiget organ, der er en privat virksomhed med autorisation fra Lægemiddelstyrelsen.
Markedsføring og sikkerhedsovervågning af medicinsk udstyr
Medicinsk udstyr skal være CE-mærket, før det kommer på markedet. CE-mærkningen viser, at udstyret opfylder den gældende EU-lovgivning. Hvis der er tale om medicinsk udstyr i en risikoklasse, der er højere end klasse I/klasse A, skal vurderingen af, om produktet kan CE-mærkes, foretages af et bemyndiget organ. Det bemyndigede organ vurderer, om dokumentationen for produktets sikkerhed og ydeevne er tilstrækkelig til, at udstyret kan CE-mærkes. Når et eventuelt certifikat er udstedt og fabrikanten har underskrevet en overensstemmelseserklæring, må fabrikanten CE-mærke sit udstyr og begynde at markedsføre det.
Når udstyret er tilgængeligt på markedet, skal fabrikanten fortsætte med at overvåge udstyret, hvilket bl.a. indebærer at have et system til at vurdere klager og indberette alvorlige hændelser til relevante kompetente myndigheder samt dokumentere dette.
Sundhedspersoner, fabrikanter, autoriserede repræsentanter, distributører og importører har pligt til at indberette alvorlige hændelser (fejl, svigt eller mangler) med medicinsk udstyr. Fabrikanter, sundhedspersoner og driftsansvarlige for offentlige og private sygehuse skal indberette direkte til den kompetente myndighed. Autoriserede repræsentanter skal indberette til fabrikanten, mens importører og distributører skal indberette til fabrikanten eller evt. til importøren eller den autoriserede repræsentant.
Borgere har også mulighed for at indberette alvorlige og formodede alvorlige hændelser til Lægemiddelstyrelsen.
Lægemiddelstyrelsen vurderer alle indberetninger for sammen med fabrikanten at afgøre, om fabrikanten f.eks. skal foretage ændringer ved udstyret, opdatere udstyrets brugsvejledning eller om udstyret i yderste konsekvens skal trækkes helt af markedet. Styrelsen samarbejder af samme årsag med myndigheder i andre lande, herunder i EU.
Løbende re-certificering af medicinsk udstyr i mellem- og højrisikoklasse
Udstyr, der tilhører mellem- og højrisikoklasse (IIa, IIb, og III/ klasse B, C og D) skal løbende re-certificeres. Det vil sige, at udstyrets dokumentation med jævne mellemrum skal gennemgås af eksperter i de bemyndigede organer. Gennemgangen af dokumentation skal ske mindst hvert femte år for at sikre, at udstyret stadig er sikkert at bruge og fungerer efter hensigten.