
What is a clinical trial?
Clinical trials are investigations of the effects and side effects of medicines. A clinical trial is conducted to find out how a medicine works, what side effects it has, and how it is metabolized in the body.
Clinical trials with medicines take place in different phases, which are overall categories of the various stages of development. Phases I, II, and III are typically trials with non-approved medicines under development. Phase I and II trials generally include relatively few participants, while Phase III can potentially include thousands. Phase IV trials involve already approved medicines, investigating the existing use of the medicine or comparable treatments.
Read more here: ICH E8 General considerations for clinical studies - Scientific guideline | European Medicines Agency (europa.eu)
The Danish Medicines Agency works to ensure that people can trust in the safety and reliability of participating in clinical trials.
Voluntary participants test medicines
Clinical trials can include either healthy, voluntary participants or voluntary patients, depending on what is being investigated. All participants in clinical trials must receive both oral and written information about the trial and give their written consent to participate before starting the trial. In special cases, other forms of consent may be used, which can be read more about at the Medical Research Ethics Committees (MREC).
Clinical trials must be approved
In Denmark, clinical trials must be approved by the Danish Medicines Agency and the Medical Research Ethics Committees (MREC) before they can begin. The overall responsible for the trial (sponsor) applies through the EU portal Clinical Trial Information System (CTIS). When the application is submitted in CTIS, it is sent to both the Danish Medicines Agency and MREC. The Danish Medicines Agency conducts a thorough assessment of the trial’s quality and the safety of the participants. MREC assesses, among other things, the ethical aspects of the trial and the scientific basis.
The Danish Medicines Agency’s Clinical Trials Unit (DKMA-CTU) coordinates a joint assessment of Part I in CTIS. Part II is assessed solely by MREC.
In Denmark, MREC is centralized at the Danish National Center for Ethics, which coordinates the ethical assessment of clinical trials. Read more on MREC’s website.
The efficient internal cooperation between MREC and DKMA-CTU provides applicants with easy access to quick and competent help both before, during, and after the approval process.
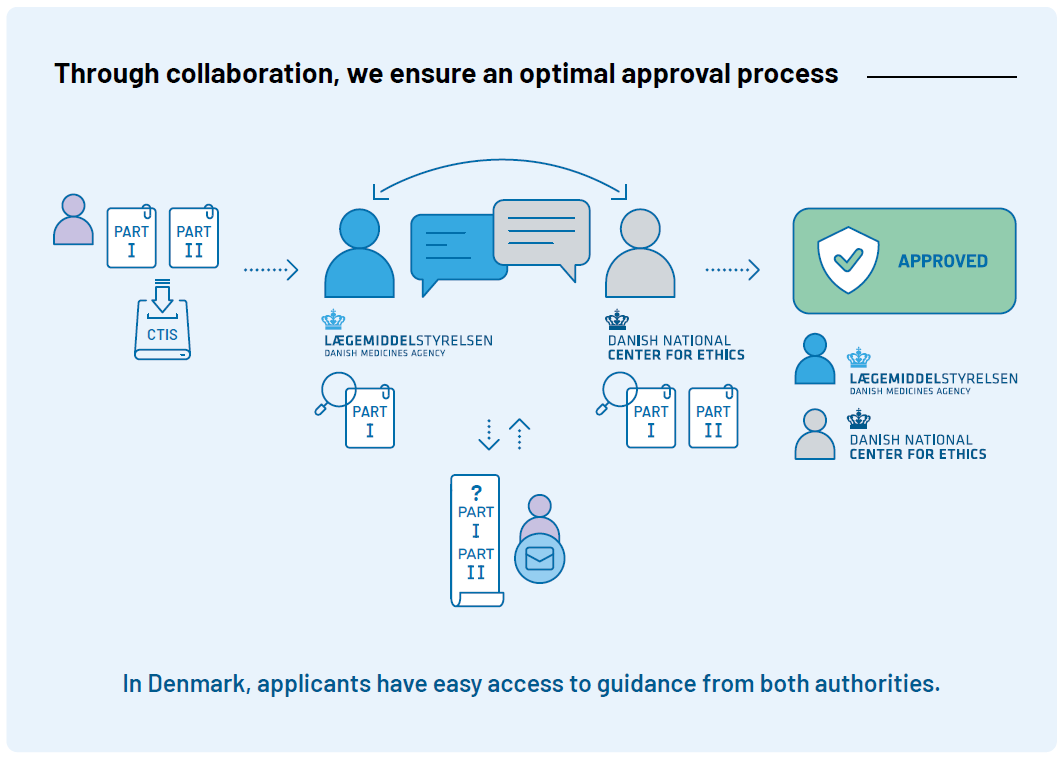
See here for a higher resolution of the approval process.
The central database for clinical trials – Clinical Trial Information System (CTIS)
CTIS is managed by the central European Medicines Agency (EMA), but it is the national member states’ medicines agencies and ethical committees that jointly assess the clinical trials.
CTIS is the central place for submission and subsequent correspondence between the authorities and the sponsor regarding the questions (considerations) the authorities may have before the trial can be approved and the sponsor’s responses. The decision is also issued in CTIS.
For trials applied for in several countries (multinational), the approval process is coordinated by one of the member states chosen as the Reporting Member State (RMS). The RMS coordinates the overall assessment, which is presented in CTIS.
It is possible for everyone to search for information about trials in Denmark and the EU in the CTIS database. You can read more about when information about clinical trials is published here.
Trials approved before January 31, 2022, must be searched in the register of the previous system (clinicaltrialsregister).
Inspection of Clinical Trials
The Danish Medicines Agency (DKMA) conducts inspections of clinical trials to ensure compliance with the approved protocol and the guidelines for Good Clinical Practice (GCP).
DKMA inspections of clinical trials with medicinal products both in Denmark and abroad, including inspections coordinated through the European Medicines Agency (EMA).
You can read more about GCP inspections here.
Adverse Events in Clinical Trials with Medicinal Products
Investigators are responsible for collecting and recording adverse events in accordance with the trial protocol. In the event of serious unexpected adverse reactions, the sponsor must immediately report these to EudraVigilance (the EU’s adverse reaction database). The Danish Medicines Agency monitors these reports in EudraVigilance. You can read more about managing and reporting adverse reactions in clinical trials here.
EU Collaboration on Clinical Trials
European medicines agencies are organized under the Heads of Medicines Agencies (HMA), which includes the Clinical Trial Coordination Group (CTCG). Denmark is highly active in this group, working to harmonize and strengthen approval procedures among member states and to develop guidance for applicants/sponsors.
There is also European collaboration under the European Medicines Agency (EMA) to promote and optimize clinical trials, even though the approval of clinical trials remains a national responsibility.
Learn more about the Accelerating Clinical Trials in the EU program here.
Questions and Answers
We have compiled a list of questions and answers about clinical trials with medicinal products, including whether a specific trial requires an application to the Danish Medicines Agency. You can find it here: Questions and Answers.
If you cannot find the answer you are looking for, you are always welcome to call us or send an email.