
Product defects and withdrawal of medicinal products in 2008
Introduction
Summary of findings in 2008
Reports
- Number of reported product defects
- Reports broken down by source
- Reports broken down by type of defect
- Counterfeit medicines
Withdrawals
- Number of withdrawals
- Withdrawals broken down by reporting source
Introduction
A company must notify the Danish Medicines Agency via reports if it considers that a product defect may lead to the withdrawal of a medicinal product from the market. This is stipulated in section 30 of Executive Order no. 1242 of 12 December 2005 on the manufacturing and import of medicinal products and intermediary products.
Via the so-called ‘Rapid Alert System’, the Danish Medicines Agency receives warnings about product defects from foreign medicines agencies and reports new product defects at international level. The Rapid Alert System covers medicinal products in the legal supply chain only.
The reports are divided into three classes: I, II and III according to the severity of the defect. Class I defects are potentially life threatening and requires that a rapid alert notification must be sent to all parties. Class II and class III defects could involve patient risk and are treated accordingly. The Danish Medicines Agency’s reports for 2008 mainly constitute class I and class II defects. In addition to the Rapid Alert System, Danish and foreign companies may report product defects to the Danish Medicines Agency directly.
The Danish Medicines Agency investigates all reports thoroughly. Before a decision is made to withdraw a medicinal product from the market, several aspects must be examined, among other things whether the medicinal product is marketed in Denmark, whether it is being clinically tested or dispensed via a special compassionate use permit. In addition, the extent to which the product defect presents a potential risk for the patient is investigated. Moreover, the medicinal product might have been exported from Denmark.
If a defective medicinal product is on the Danish market, or if it has been exported, the Danish Medicines Agency may effect a withdrawal in collaboration with the company. It is estimated how far down the supply chain the medicinal product needs to be withdrawn (wholesaler, pharmacy, consumer), and whether it is relevant to inform other medicines agencies about the withdrawal. Where critical product defects are concerned, we place warnings on our website, www.dkma.dk. Where less critical product defects are concerned, we also consider how a withdrawal of the medicinal product from the market would effect the supply of medicine to the Danish people.
Companies must comply with the rules on good manufacturing and distribution practice (GMP and GDP). The rules contribute to minimising potential defects during the manufacturing process and distribution of medicinal products. At inspections, we check whether companies comply with these rules. In addition, we regularly purchase selected medicinal products, packages, labels and analyses for laboratory testing at the Danish Medicines Agency. During case handling of variations of marketing authorisations, submitted information might uncover defects or changes in medicinal products. Withdrawal of medicinal products could therefore also be effected as a result of the Danish Medicines Agency’s own testing and authorisation of medicinal products or in connection with company inspections.
Summary of findings in 2008
In 2008, 177 reports of medicinal product defects were registered, which is less than in 2007. 23% of these reports led to products being withdrawn from the market, which is also less than in the preceding years. Most of the withdrawals effected in 2008 were occasioned by reports made by the companies. The most frequent reason for withdrawal was ‘packaging defects’.
Reports
Number of reported product defects
In 2008, we received a total of 177 reports of medicinal product defects. Figure 1 shows the number of reports received in the period 2005-2008. The number of submitted reports fell in 2008 compared with the preceding years during which period the volume had been rising.
Fig. 1. Figure 1. Number of reports of product defects 2005-2008
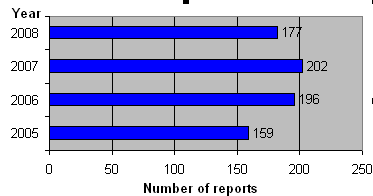
The 177 reports of product defects in 2008 both concern marketed and non-marketed medicinal products, and cover
- reports of product defects from companies and authorities (> 90 % of the reports)
- complaints from medicine users
- discovery of counterfeit medicines in the legal supply chain not manufactured by the authorised pharmaceutical manufacturer, and where quality, effect and safety are not documented
- reports of companies’ non-compliance with GMP or GDP, uncovered from company inspections carried out by European medicines agencies.
The reporting sources of the 177 reports submitted to the Danish Medicines Agency in 2008 and 2007 appear from figure 2.
Figure 2. Reports broken down by source
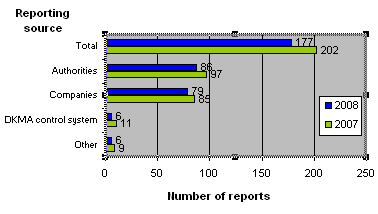
2008 generally saw fewer product defect reports. The Danish Medicines Agency's control and licensing units have recorded a decline in submitted reports compared with 2007. Likewise, the year recorded a small decrease in the number of reports submitted by companies (manufacturers and importers) and from other medicines agencies compared with 2007. In 2008, a slightly lower number of reports came from other sources (patients, physicians and pharmacies) compared with 2007.
Reports broken down by type of defect
When the reports are registered at the Danish Medicines Agency, they are divided into six different types of defects.
Figure 3 compares reports received in 2007 and 2008, broken down by source. Box 1 briefly describes each of the types of defects.
Figure 3. Reports broken down by type of defect
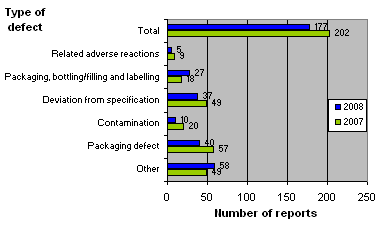
Compared with 2007, 2008 recorded an increase in defects in the category “Packaging, bottling/filling and labelling" and an increase in the category “Other”. By contrast, the number of reported defects related to adverse reactions, deviations from specifications, contamination and packaging defects fell on 2007.
|
Box. 1. Types of defects
|
In the period 2003-2007, the Rapid Alert System transmitted an increasing number of warnings about counterfeit medicines found in the legitimate supply chain globally, cf. table 1. Such warnings decreased decisively in 2008, cf. table 1. The Danish Medicines Agency has not received information about or observed any counterfeit medicines in the legitimate supply chain in Denmark so far.
The decline in the number of reports submitted in 2008 may be ascribed to a smaller number of recorded counterfeit products at global level and a general fall in the number of reports submitted to the Danish Medicines Agency.
Table 1. Number of reports of counterfeit medicines in the period 2003-2008.
|
|
2003 |
2004 |
2005 |
2006 |
2007 |
2008 |
|
Counterfeit medicines |
0 |
2 |
3 |
8 |
19 |
3 |
As part of our activities to prevent counterfeit medicines from reaching consumers, the Danish Medicines Agency has formed a network against counterfeit medicines. The network is made up of authorities, industrial associations and all links in the supply chain. The network participants meet biannually to discuss issues related to counterfeit medicines and to initiate local and cross-cutting preventive measures accordingly.
The network is presently drafting guidelines the objective of which is to help pharmaceutical companies and pharmacies in their efforts to prevent counterfeit medicines from entering the legal supply chain.
In the near future, it will be possible for companies to report information about counterfeit medicines via an e-form at the Danish Medicines Agency’s website. This will facilitate the reporting procedures for companies covered by the reporting duty laid down in section 43b of the Danish Medicines Act (Act no. 1180 of 12 December 2005). The reporting duty was implemented by amendment of the Danish Medicines Act on 1 July 2008 and obligates marketing authorisation holders pursuant to section 7 and holders of a company authorisation pursuant to section 39 (1) to report any discovery of counterfeit medicines to the Danish Medicines Agency.
The Danish Medicines Agency participates in the European cooperation between medicines agencies in the EU which exchanges information about illegal, including counterfeit, medicinal products distributed in the illegal supply chain in Europe. The information exchanged could concern pharmaceuticals sold illegally via the internet, which could be related to a multitude of countries due to the global nature of the internet. The Danish Medicines Agency investigates such information if it seems to be related to Denmark and sanctioning measures are required. In addition we publish warnings on our website whenever it is necessary to inform Danish consumers. In circumstances that may be penalised under the Danish Medicines Act, the Danish Medicines Agency takes decisions regarding the liable party and refers, according to the circumstances, such cases to the police for imposition of legal penalties for the violation of the provisions of the Danish Medicines Act.
Withdrawals
From the total of 177 product defect reports received in 2008, 23% resulted in actual withdrawals of medicinal products from the Danish market.
The total number of reports has dropped compared with the two preceding years, 2006-2007, cf. table 2. Furthermore, the number of withdrawals, also as a percentage of the total number of reports, has declined compared with the four preceding years, 2004-2007.
Table 2. Number of withdrawals in the period 2002-2008.
|
|
2002 |
2003 |
2004 |
2005 |
2006 |
2007 |
2008 |
|
Number of reports |
192 |
150 |
143 |
160 |
196 |
202 |
177 |
|
Number of withdrawals |
72 |
39 |
58 |
58 |
55 |
57 |
41 |
|
Withdrawals in per cent |
38% |
26% |
41% |
36% |
28% |
28% |
23% |
Withdrawals broken down by reporting source
Table 3 shows the distribution of the 41 withdrawals in 2008 on reporting source.
Table 3. Reports resulting in withdrawals broken down by reporting source in 2006-2008
|
|
Number in 2006 |
Number in 2007 |
Number in 2008 |
|
Company |
39 |
47 |
29 |
|
Foreign authority |
10 |
4 |
8 |
|
Danish Medicines Agency’s control system |
2 |
5 |
3 |
|
Other |
4 |
1 |
1 |
|
I alt |
55 |
57 |
41 |
In 2008, a relatively smaller number of withdrawals were occasioned by company reports compared with 2007. This is also the case for withdrawals resulting from product defects notified via the Danish Medicines Agency's control system. By contrast, 2008 saw a higher number of withdrawals reported by foreign authorities than was the case in 2007. The category ‘Other’ (patients, physicians, pharmacies) is unchanged compared with 2007.
The 41 withdrawals effected in 2008 were primarily caused by ‘packaging defects’. Moreover, product defects in the categories ‘other deviations’ and ‘packaging, bottling/filling and labelling’ accounted for a large part of the withdrawals.
Some 10% of the withdrawals were withdrawn from parallel importers/distributors.
For further information, please contact Charlotte Henriksen, tel. +45 44 88 92 51.
Danish Medicines Agency, 16 April 2009