
Safety features - handling of alerts
Handling of alerts in the repositories system enters into force on 9 February 2019 under the safety feature provisions
The safety feature provisions enter into force on 9 February 2019 and aim to prevent falsified medicinal products from entering the legal pharmaceutical supply chain. The new safety requirements for the packaging of medicinal products and a new pan-European repositories system (medicinal products database) will make it possible to identify and verify the authenticity of the medicinal products comprised by the rules.
As part of the new safety requirements, the pharmaceutical manufacturer will be required to place safety features on the packaging of the respective medicinal products, namely a unique identifier in the form of a two-dimensional barcode as well as an anti-tampering device. The two-dimensional barcode must contain information on the product code, serial number, batch number and expiry date. A pan-European repositories system has also been established to which the pharmaceutical manufacturer must upload the information from the two-dimensional barcode before the product is released for sale or distribution. Finally, pharmacies and hospital pharmacies must verify the authenticity of the medicinal products by reading the two-dimensional barcode and retrieve the unique identifier and decommission the medicine in the repositories system while ensuring that the anti-tampering device is intact when the medicine is handed out to the patient.
The repositories system has been constructed by the European Medicines Verification Organisation (EMVO) in collaboration with corresponding national organisations. The Danish Medicines Verification Organisation (DMVO) is the corresponding organisation in Denmark, and it is the pharmaceutical manufacturers that must establish and operate the repositories system. The repositories system contains records of all actions related to the unique identifier, including information about which users have undertaken these actions, and more specifically the nature of those actions. The national competent authorities, i.e. the Danish Medicines Agency in Denmark, must have access to the repositories system, primarily for the purpose of investigating suspected falsification. The Danish Medicines Agency will also be supervising the operation of the repositories system. The system is used as an end-to-end verification of the medicinal products.
Duty to report
Marketing authorisation holders, manufacturers, parallel importers, parallel distributors, wholesalers, pharmacies and hospital pharmacies have a duty to report any findings of medicinal products that are or may be falsified medicinal product packs to the Danish Medicines Agency. The purpose is to enable the Danish Medicines Agency to ensure that a relevant and effective investigation of the potential falsification will take place, including an investigation of any violations of the rules and an appropriate delimitation of the damage, which could potentially result in for example the withdrawal of one or several batches of medicinal products. As part of such investigations, the Danish Medicines Agency will be in contact with the involved companies and authorities in other countries as required.
Suspected falsification
A falsification of a medicinal product could be suspected if a pack looks suspicious or if the packaging of a medicinal product pack has been tampered with. With the new system, suspicion could also be raised if a medicinal product pack cannot be verified in the repositories system, and an alert is triggered by the system, and a technical/procedural error is subsequently ruled out. If a falsification is suspected, the medicinal product must not be delivered, exported or handed out to the public.
Alert triggered by verification and attempt to decommission the medicinal product pack in the repositories system
If an alert is triggered, thus implying that a medicinal product pack must not be handed out to the patient, exported or otherwise supplied to the public, it is important to check if the alert was triggered by a technical/procedural error. This check is done at the place where the alert was triggered and subsequently at the marketing authorisation holder/manufacturer. If a technical/procedural error is ruled out, it is a case of suspected falsification to be reported to the Danish Medicines Agency.
Process description – the roles involved when an alert is triggered
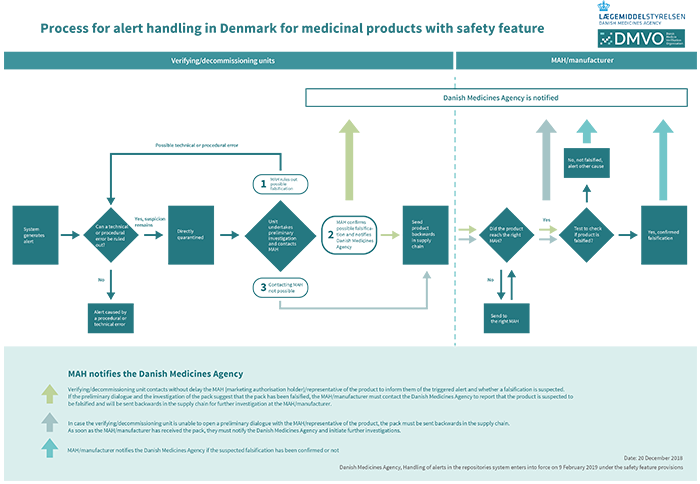
Click for larger image
In the event that a medicinal product to be handed out to a patient cannot be verified in the repositories system and an alert is triggered, the pack in question must not be handed out to the patient.
The pharmacy/hospital pharmacy must without delay check if the alert was caused by a technical/procedural error at the pharmacy/hospital pharmacy. The pharmacy/hospital pharmacy only has access to data on technical/procedural errors occurring while the medicinal product pack was in their custody. The hospital/hospital pharmacy does not have data access to investigate technical/procedural errors that may have occurred earlier in the process, e.g. at the pharmaceutical manufacturer or at the marketing authorisation holder.
If the pharmacy/hospital pharmacy’s preliminary investigation rules out that the alert was caused by a technical/procedural on their part, the medicinal product pack must be examined further to see if it is falsified. In such a situation, the pharmacy/hospital pharmacy must without delay quarantine the medicinal product pack and keep it separate from other sellable medicinal products. The pharmacy/hospital pharmacy must without delay contact the marketing authorisation holder/representative of the product to notify them about the alert triggered by the medicinal product pack. If the pack is still suspected to be a falsification after the preliminary dialogue, the pack must without delay be returned to the wholesaler having supplied the pack. In case it is not possible to open a preliminary dialogue with the marketing authorisation holder/representative of the product, the pack must be returned to the marketing authorisation holder/manufacturer via the wholesaler.
If the verifying unit in the form of e.g. the wholesaler discovers a status in the repositories system that will trigger an alert in the system later on, that alert must be responded to in the same way as described above.
Note!
Exempted are medicinal products that trigger a date mismatch alert (#52). This type of alert is triggered because the manufacturer/parallel importer has uploaded conflicting information to EMVS in the date field. Due to the extent and since the error is known, the #A52 alert must be suppressed until the error has been corrected. MAHs are not to start an investigation, but must ensure data are corrected in the hub at the earliest opportunity.
The role of the wholesaler:
The wholesaler receives the suspected medicinal product pack from the pharmacy. The suspected medicinal product pack must immediately be quarantined and kept separate from other sellable medicinal products.
The wholesaler must, within a short period of time, clarify if a technical/procedural error can explain the alert. However, the wholesaler owns only the data related to his own verification.
If a technical/procedural error is ruled out, the wholesaler returns the pack to the marketing authorisation holder/manufacturer for further testing of the suspected pack.
The role of the marketing authorisation holder/manufacturer:
The marketing authorisation holder/manufacturer is obliged to have a procedure in place for the receipt and processing of a reported complaint, product defect or suspected falsification with a 24/7 preparedness as required by the situation.
It is usually only the marketing authorisation holder/manufacturer who can positively verify if the medicinal product pack has been falsified because it requires detailed knowledge of the medicinal product to compare the suspected pack with the packs the marketing authorisation holder has put on the markets.
The pack is received by the marketing authorisation holder/manufacturer, who investigates and tests the pack, ultimately establishing if it is a technical/procedural error or if the pack is in fact a falsification.
If it is a parallel imported/parallel distributed medicinal product, the pack is returned to the parallel importer/parallel distributor who is the marketing authorisation holder of the product. If further clarification is needed, the product is hereafter sent to the original manufacturer for further investigations.
The marketing authorisation holder/manufacturer must report packs to the Danish Medicines Agency if a dialogue with the verifying/decommissioning unit has taken place, and if the preliminary investigations of the pack suggest that the pack has been falsified, implying that the pack must be returned for further investigations.
The marketing authorisation holder/manufacturer must report packs immediately to the Danish Medicines Agency if they – possibly after thorough investigations – confirm the medicinal pack to be falsified.
If the marketing authorisation holder/manufacturer concludes that a pack that was previously reported to the Danish Medicines Agency as a suspected falsification is not a falsification, the Danish Medicines Agency must be notified.
Anti-tampering device (ATD)
If the anti-tampering device on a medicinal product pack has been broken, then follow the process as described under triggered alert.
Medicines dispensed according to a compassionate use permit
Medicinal products authorised in the EU and released for sale and distribution in the EU are covered by the requirements, and the safety features must therefore be added in the country where they are marketed. This means that medicinal products supplied according to a compassionate use permit but marketed in another EU country than Denmark are covered by the safety feature provisions.
Medicinal products that are not authorised in the EU but authorised outside the EU and supplied according to a compassionate use permit are exempt from the safety feature provisions.
Reporting to the Danish Medicines Agency
Marketing authorisation holders/manufacturers are responsible for reporting medicinal products that are or may have been falsified to Send an email.
Medicinal products released before 9 February 2019
Because the safety feature provisions do not enter into force before 9 February 2019, medicinal products released before this date may not necessarily be uploaded to the repositories system. Attempts to verify these products will appear in the system as "unknown". As medicinal products may have a shelf life of up to five years, it may take up to five years before all medicinal product packs covered by the safety feature provisions are uploaded in the repositories system. The Danish Medicines Agency finds that during this period, no action should be taken in response to medicinal products returned as "unknown" by the repositories system in connection with a verification attempt. The suppression of packs returned as "unknown" by the system has to do with the product's authorised shelf life. It remains important, however, to report it to the Danish Medicines Agency if a medicinal product pack looks suspicious or if there are visible signs of falsification.
Medicinal products released after 9 February 2019
If the marketing authorisation holder/manufacturer by mistake fails to upload data to the system, the pack will also appear as "unknown" in the system. In such a situation, we only expect a report to be made to the Danish Medicines Agency if the pack has visible signs of falsification. A pack that has been released after 9 February must not be delivered, exported or handed out to the public if the pack appears as "unknown" in the system and thus remains to be uploaded to the repositories system. It is indicated in the medicine price files (the price list of medicinal productes) which medicines are covered by the safety feature provisions as well as when a medicine's approved shelf life indicates that the medicinal product packs should be uploaded to the repositories system.