
Sikkerhedselementer - håndtering af alarm
Håndtering af alarmer fra lægemiddeldatabasen efter reglerne om sikkerhedselementer træder i kraft 9. februar 2019
Reglerne om sikkerhedselementer træder i kraft den 9. februar 2019 og skal forhindre, at der kommer forfalskede lægemidler ind i den legale forsyningskæde for lægemidler. Med de nye sikkerhedskrav til lægemidlers emballage og en fælles europæisk lægemiddeldatabase bliver det muligt at identificere og kontrollere ægtheden af de lægemidler, der er omfattet af reglerne.
Med de nye regler om sikkerhedselementer indføres der bl.a. krav om, at lægemiddelfremstilleren skal forsyne de omfattede lægemidlers emballage med sikkerhedselementer, nemlig en entydig identifikator i form af en 2D-stregkode og en anbrudsanordning. 2D-stregkoden skal indeholde oplysning om produktkode, serienummer, batchnummer og udløbsdato. Samtidig er der oprettet en fælles europæisk lægemiddeldatabase, hvori lægemiddelfremstilleren skal uploade informationer fra 2D-stregkoden, inden lægemidlet frigives til salg eller distribution. Endelig skal apotekerne og sygehusapotekerne verificere lægemiddels ægthed ved at aflæse 2D-stregkoden og genfinde den entydige identifikator og deaktivere lægemidlet i databasen, ligesom det skal kontrolleres, om anbrudsanordningen er intakt i forbindelse med, at lægemidlet udleveres til patienten.
Lægemiddeldatabasen er blevet opbygget af European Medicines Verification Organisation (EMVO) i samarbejde med tilsvarende nationale organisationer, i Danmark findes tilsvarende Dansk Medicin Verifikation Organisation (DMVO), da det er lægemiddelfremstillerne, der skal oprette og drive databasen. Databasen indeholder fortegnelser over alle handlinger vedrørende en entydig identifikator, herunder oplysninger om, hvilke brugere der har forestået disse handlinger, og nærmere hvad handlingerne bestod i. De nationale kompetente myndigheder, dvs. i Danmark Lægemiddelstyrelsen, skal have adgang til databasen, primært med henblik på efterforskning af mistanke om forfalskning. Lægemiddelstyrelsen skal også føre tilsyn med driften af databasen. Databasen bruges som en start-til-slut kontrol af lægemidlerne.
Pligt til at indberette
Markedsføringsindehavere, fremstillere, parallelimportører, paralleldistributører, grossister, apoteker og sygehusapoteker har pligt til at indberette fund af lægemidler, som er eller kan være forfalskede lægemiddelpakninger til Lægemiddelstyrelsen. Formålet med dette er, at Lægemiddelstyrelsen kan sikre, at der sker en relevant og effektiv undersøgelse af den mulige forfalskning, herunder en efterforskning af en eventuel overtrædelse af reglerne og en relevant afgræsning af skaden, hvilket f.eks. kan betyde tilbagekaldelser af en eller flere batches af et lægemiddel. I forbindelse med en sådan sagsbehandling er Lægemiddelstyrelsen i kontakt med involverede virksomheder, og eventuelt myndigheder i andre lande.
Mistanke om forfalskning
Mistanke om at et lægemiddel er potentielt forfalsket kan opstå, hvis en pakning ser mistænkelig ud eller, hvis emballagen af en lægemiddelpakning er blevet brudt. Med det nye system kan det også være, hvis en lægemiddelpakning ikke kan verificeres i lægemiddeldatabasen, og en alarm udløses af systemet, og det efterfølgende kan udelukkes, at der er tale om en teknisk-/procedurefejl. Hvis der er mistanke om en forfalskning, må lægemidlet ikke leveres, eksporteres eller udleveres til offentligheden.
Alarm udløst ved verificering og forsøg på deaktivering af lægemiddelpakning i lægemiddeldatabasen
Hvis en alarm (alert) udløses, og en lægemiddelpakning dermed ikke må udleveres til en patient, eksporteres eller udleveres til offentligheden i øvrigt, er det vigtig at undersøge om alarmen er udløst pga. en teknisk-/procedurefejl. Denne undersøgelse foretages hvor alarmen er udløst og derefter hos markedsføringsindehaveren/ fremstilleren. Hvis en teknisk-/procedurefejl kan udelukkes, vil der være tale om en mistanke om forfalskning, som skal indberettes til Lægemiddelstyrelsen.
Procesbeskrivelse af aktørers rolle ved udløst alarm
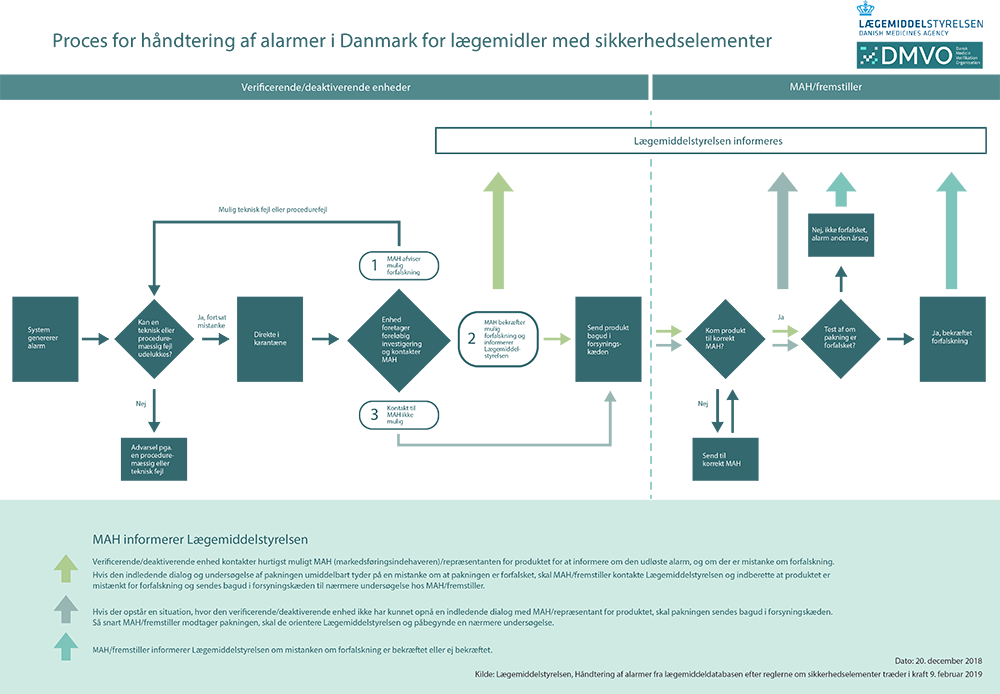
Klik for stort billede (pdf i A3-størrelse)
Verificerende/deaktiverende enheds rolle:
Hvis en lægemiddelpakning ved tiltænkt udlevering til en patient ikke kan verificeres i lægemiddeldatabasen, og en alarm udløses, må lægemiddelpakningen ikke udleveres til patienten.
Apoteket/sygehusapoteket skal hurtigst muligt undersøge, om alarmen kan skyldes en teknisk-/procedurefejl sket på apoteket/sygehusapoteket, som kan forklare alarmen. Apoteket/sygehusapoteket har kun adgang til data om teknisk-/procedurefejl, der er opstået, mens lægemiddelpakningen har været i deres forvaring. Teknisk-/procedurefejl som er opstået længere tilbage i processen hos f.eks. lægemiddelfremstilleren eller markedsføringsindehaveren, har apoteket/sygehusapoteket ikke dataadgang til at undersøge.
Hvis apotekets/sygehusapotekets indledende undersøgelse kan udelukke, at alarmen skyldes en teknisk-/procedurefejl sket på apoteket/sygehusapoteket, skal lægemiddelpakken undersøges nærmere for forfalskning. Apoteket/sygehusapoteket skal i en sådan situation hurtigst muligt sætte den mistænkte lægemiddelpakning i karantæne, samt holde pakningen adskilt fra andre salgbare lægemidler. Apoteket/sygehusapoteket skal hurtigst muligt kontakte markedsføringsindehaveren/repræsentanten for produktet, for at informere om den udløste alarm på lægemiddelpakningen. Hvis pakningen efter indledende dialog stadig er mistænkt, skal pakningen hurtigst muligt sendes retur til den grossist, som har leveret pakningen. Hvis der opstår en situation, hvor det ikke er muligt at opnå en indledende dialog med markedsføringsindehaver/repræsentanten for produktet, skal pakningen sendes retur til markedsføringsindehaver/ fremstiller via grossisten.
Hvis verificerende enhed i form af f.eks. grossisten opdager en status i lægemiddeldatabasen, som senere i systemet vil udløse en alarm, skal der tages aktion på denne advarsel på samme måde som ovenfor beskrevet.
OBS:
Undtaget er lægemidler, hvor alarmen udgør dato mismatch (#A52). Alarmtypen skyldes, at producent/parallelimportør har uploaded uoverensstemmende information til EMVS i dato-feltet, hvilket udløser alarmen. Grundet omfanget og idet fejlen er kendt, må #A52 frem til denne fejl er udbedret, undtrykkes, og MAH'er skal ikke påbegynde en undersøgelse, men snarest sikre, at data rettes til på hub'en.
Grossistens rolle:
Grossisten modtager den mistænkte lægemiddelpakning retur fra apoteket. Den mistænkte lægemiddelpakning skal straks sættes i karantæne samt holdes adskilt fra øvrige salgbare lægemidler.
Grossisten skal inden for kort tid afklare, om grossisten kan finde en teknisk-/procedurefejl, der kan forklare alarmen. Grossisten ejer dog kun data fra deres egen verificering.
Hvis en teknisk-/procedurefejl kan udelukkes sender grossisten pakningen retur til markedsføringsindehaveren/ fremstilleren til nærmere testning af den mistænkte pakning.
Markedsføringsindehavers/ fremstillers rolle:
Markedsføringsindehaver/ fremstiller er forpligtet til at have en procedure for at kunne modtage og behandle en indberetning om reklamation, produktfejl eller mistanke om forfalskning med et 24-7 beredskab, hvis nødvendigt.
Det vil normalt kun været markedsføringsindehaver/ fremstiller, der positivt kan konstatere, om lægemiddelpakningen er forfalsket, da dette kræver en nøjere viden om lægemidlet, som gør det muligt at sammenligne pakningen med de pakninger, som markedsføringsindehaver har sendt på markederne.
Pakning modtages af markedsføringsindehaver/ fremstiller, som undersøger og tester pakningen, hvor udfaldet kan være en teknisk/procedurefejl eller bekræftelse af, at pakning er forfalsket.
Ved parallelimporteret/paralleldistribueret lægemiddel sendes pakningen til den parallelimportør/paralleldistributør, som er markedsføringsindehaver for produktet. Hvis der er behov for nærmere afklaring, sendes produkt herefter til originalfremstiller til nærmere afklaring.
Markedsføringsindehaver/fremstiller skal indberette pakninger til Lægemiddelstyrelsen, hvor der har været en dialog med verificerende/deaktiverende enhed, og hvor en indledende undersøgelse af pakningen umiddelbart tyder på en mistanke om at pakningen er forfalsket, og det dermed er nødvendigt at få pakningen retur til nærmere undersøgelse.
Markedsføringsindehaver/fremstiller skal indberette pakninger straks til Lægemiddelstyrelsen, hvis de – eventuelt efter en grundigere undersøgelse – bekræfter at lægemiddelpakningen er forfalsket.
Hvis markedsføringsindehaver/fremstiller konkluderer, at en pakning som tidligere har været indberettet til Lægemiddelstyrelsen som mistænkt forfalsket, ikke er forfalsket, skal Lægemiddelstyrelsen underrettes herom.
Anbrudsordning (ATD)
Hvis anbrudsordningen på en lægemiddelpakning er brudt, følges samme procesbeskrivelse som beskrevet under udløst alarm.
Lægemidler på udleveringstilladelse
Lægemidler der er godkendt inden for EU, og som frigives til salg og distribution i EU, er omfattet af kravene og skal derfor påføres sikkerhedselementer i det land, som lægemidlerne markedsføres i. Det betyder, at lægemidler der er givet på udleveringstilladelse i Danmark, men som er markedsført i et andet EU-land end Danmark, er omfattet af reglerne om sikkerhedselementer.
Lægemidler der ikke er godkendt i EU, men som er godkendt uden for EU, og som er givet på udleveringstilladelse, er undtaget for reglerne om sikkerhedselementer.
Indberetning til Lægemiddelstyrelsen
Markedsføringsindehaver/ fremstiller har ansvaret for at indberette et lægemiddel, der er eller kan være forfalsket til Lægemiddelstyrelsen via nedenstående link:
Indberetning af lægemiddelpakning(er) der er mistænkt forfalsket
Bemærk at udfyldelsen af formularen aldrig må forsinke indberetning af potentielle forfalskninger til os. I så fald bør du kontakte os hurtigst muligt via telefon 44 88 95 95 eller via e-mail Send en mail og videregive de foreliggende informationer (mail er bemandet i Lægemiddelstyrelsens åbningstid)
Lægemidler frigivet før 9. februar 2019
Da reglerne om sikkerhedselementer først træder i kraft den 9. februar 2019, vil lægemidler frigivet før denne dato ikke nødvendigvis være uploadet i lægemiddeldatabasen, og hvis disse forsøges verificeret, vil de fremgå i systemet som værende ”ukendte”. Da lægemidler kan have op til 5 års holdbarhed, vil der være en periode på op til 5 år før alle lægemiddelpakninger, som er omfattet af reglerne ift. sikkerhedselementer, vil være uploadet i lægemiddeldatabasen. Det er Lægemiddelstyrelsens holdning, at der i denne periode ikke skal reageres ud fra lægemiddeldatabasens oplysninger om lægemiddelpakninger, der ved forsøg på verificering, angives som ”ukendte” i systemet. Undertrykkelsen af pakninger som i systemet fremgår som værende ”ukendte” er afhængig af produktets godkendte holdbarhed. Det er dog stadig vigtigt at indberette til Lægemiddelstyrelsen, hvis en lægemiddelpakning er mistænkelig eller viser synlige tegn på forfalskning.
Lægemidler frigivet efter 9. februar 2019
Hvis markedsføringsindehaver/ fremstiller ved en fejl ikke får uploadet data i systemet, vil pakningen ligeledes fremgå som ”ukendt”. I en sådan situation forventes det kun, at der indberettes til Lægemiddelstyrelsen hvis pakningen viser synlige tegn på forfalskning. En pakning som er frigivet efter 9. februar 2019 må ikke leveres, eksporteres eller udleveres til offentligheden, hvis pakningen fremgår som ”ukendt” og dermed mangler at blive uploaded i lægemiddeldatabasen. Der er markeringer i medicinprisfilerne (”taksten”), som giver et overblik over hvilke lægemidler, der er omfattet af reglerne for sikkerhedselementer samt hvornår et lægemiddels godkendte holdbarhed indikerer, at lægemiddelpakningerne bør være uploaded i lægemiddeldatabasen.