
Anmeldelsespligt
Anmeldelsespligten under de forskellige artikler i forordningen afhænger af, om udstyret er CE-mærket eller ej, og om der er intention om at anvende de indsamlede data til en overensstemmelsesvurdering af udstyret.
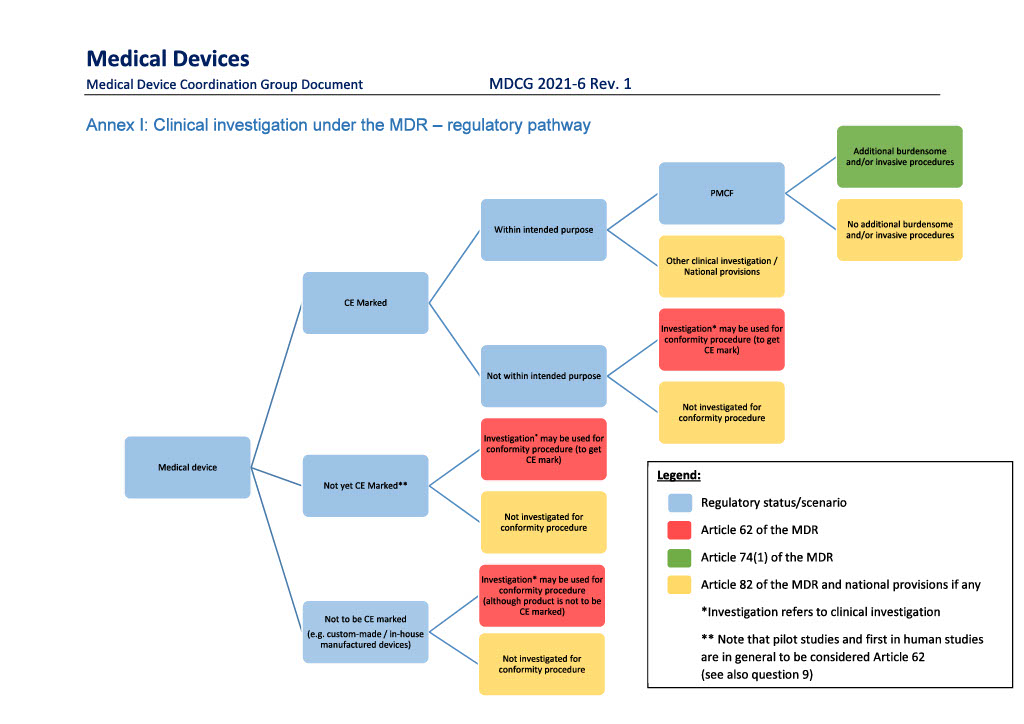
Der er 3 forskellige typer af kliniske afprøvninger af medicinsk udstyr angivet i MDR:
Artikel 62 ”Produktudvikling”: Afprøvning af medicinsk udstyr for at vurdere sikkerhed og ydeevne af udstyret, enten på ikke-CE-mærket medicinsk udstyr, eller på CE-mærket medicinsk udstyr, hvor fabrikanten ønsker at udvide produktets formål.
Artikel 74(1) ”PMCF-afprøvning med yderligere invasive/byrdefulde procedurer”: Fabrikantinitierede Post-Market kliniske opfølgningsstudier af CE-mærket udstyr, hvor forsøgspersoner udsættes for yderligere invasive/byrdefulde procedurer.
Artikel 82 ”Andre afprøvninger”.
Nedenstående figur er fra EU-Kommissionens vejledning ”MDCG 2021-6 Rev. 1 Regulation (EU) 2017/745 – Questions & Answers regarding clinical investigation”.
Vejledningen findes her.

Artikel 62 - Produktudvikling
For ikke-CE-mærket medicinsk udstyr gælder, at såfremt den kliniske afprøvning udføres som en del af den kliniske evaluering af udstyret med henblik på en efterfølgende overensstemmelsesvurdering (en proces til påvisning af, om forordningens krav vedrørende et udstyr er blevet opfyldt), med et eller flere af følgende formål:
- fastslå og verificere, at et udstyr er egnet til at opfylde et eller flere af udstyrets specifikke medicinske formål, og opnå udstyrets ydeevne,
- fastslå og verificere de kliniske fordele,
- fastslå og verificere udstyrets kliniske sikkerhed og at fastslå eventuelle uønskede bivirkninger, samt vurdere om det udgør en acceptabel risiko set i forhold til de fordele, der kan opnås med udstyret,
skal afprøvningen tillades af Lægemiddelstyrelsen, inden den kliniske afprøvning kan igangsættes.
I Danmark er der indført nationale særregler, som fastsætter, at kravet om at indhente tilladelse til en klinisk afprøvning gælder uanset udstyrets risikoklasse.
Såfremt der er tale om en klinisk afprøvning, hvor et CE-mærket udstyr anvendes udenfor udstyrets erklærede formål, og hvor afprøvningen foretages med intention om at udvide eller ændre udstyrets erklærede formål, skal afprøvningen anmeldes under artikel 62.
Ansøgning om tilladelse til en klinisk afprøvning under artikel 62 skal sendes samtidigt til både Lægemiddelstyrelsen og De Videnskabsetiske Medicinske Komitéer som én ansøgning. Du kan finde det fælles ansøgningsskema her.
Artikel 74(1) - PMCF-afprøvning med yderligere invasive eller byrdefulde procedurer
For en klinisk afprøvning af CE-mærket udstyr, hvor det medicinske udstyr anvendes i henhold til udstyrets erklærede formål, og den kliniske afprøvning er en del af fabrikantens post-market clinical follow-up (PMCF)-forpligtelser, og hvor forsøgspersonerne udsættes for yderligere byrdefulde/invasive procedurer, skal afprøvningen vurderes af De Videnskabsetiske Medicinske Komitéer under artikel 74, stk. 1. Afprøvningen skal meddeles 30 dage inden start til Lægemiddelstyrelsen. Læs mere her om, hvordan sponsor sender meddelelse til Lægemiddelstyrelsen.
Afprøvninger, hvor et CE-mærket udstyr afprøves i henhold til udstyrets formål som et led i fabrikantens PMCF-plan, kan f.eks. være afprøvninger, hvor man ønsker at indsamle kliniske data for at se effekt eller bivirkninger, når udstyret anvendes over en længere periode, og hvor der indgår ekstra byrdefulde/invasive procedurer (fx biopsi e.l.), som normalt ikke ville være udført under normal klinisk brug af udstyret.
Hvorvidt yderligere procedurer er belastende for forsøgspersoner, skal vurderes ud fra forsøgspersonens perspektiv. Belastende ekstra procedurer kan være ekstra procedurer, som giver øget risiko for komplikationer eller bivirkninger, jf. MDCG 2021-6 Regulation (EU) 2017/745 – Questions & Answers regarding clinical investigation.
Ansøgning om tilladelse til en klinisk afprøvning under artikel 74(1) skal ansøges hos De Videnskabsetiske Medicinske Komitéer og Lægemiddelstyrelsen skal underrettes om afprøvningen 30 kalenderdage, inden afprøvningen påbegyndes.
Artikel 82 - Andre afprøvninger
Såfremt den kliniske afprøvning ikke udføres med nogen af de ovenstående formål, er afprøvningen omfattet af bestemmelserne i artikel 82, der angiver en række minimumskrav for andre typer af kliniske afprøvninger af medicinsk udstyr.
Bestemmelsen i artikel 82, stk. 1 nævner bl.a., at der skal være en sponsor for afprøvningen, afprøvningen skal have en positiv udtalelse fra De Videnskabsetiske Medicinske Komitéer, forsøgspersoner skal beskyttes og give samtykke til deltagelse, og fabrikanten af udstyret skal dokumentere, at udstyret opfylder de generelle krav til sikkerhed og ydeevne listet i bilag I i forordningen undtagen for de aspekter, der er omfattet af den kliniske afprøvning.
Ansøgning om tilladelse til en klinisk afprøvning under artikel 82 skal ansøges hos De Videnskabsetiske Medicinske Komitéer.