
Produkter uden medicinsk formål
Der findes en række produkter, som minder om medicinsk udstyr, men som ikke har et medicinsk formål. Det kan f.eks. være farvede kontaktlinser uden styrke, fillers eller laser til fjernelse af tatoveringer. Fra den 22. juni 2023 er det et lovkrav, at dansketablerede fabrikanter, importører og distributører m.fl. skal være registreret hos Lægemiddelstyrelsen for at kunne markedsføre produkter uden et medicinsk formål i Danmark.
MDR Bilag 16På denne temaside fortæller vi bl.a. mere om, hvad produkter uden medicinsk formål er, hvordan man registrerer sig, og hvordan man skal indberette hændelser med produkterne.
Hvad er produkter uden medicinsk formål?
Der findes en række produkter, som minder om medicinsk udstyr, men som ikke har et medicinsk formål. Disse produkter er ikke tidligere faldet ind under definitionen for medicinsk udstyr og dermed reglerne i EU-forordningen for medicinsk udstyr.
Af hensyn til patientsikkerheden og for at sikre, at disse produkter opfylder en række krav til sikkerhed og ydeevne, bliver de nu omfattet af EU-forordningen for medicinsk udstyr (MDR).
Produkterne fremgår i forordningens bilag 16 og omfatter for nuværende bl.a. følgende produktgrupper:
- Kontaktlinser uden styrke, f.eks. farvede kontaktlinser
- Produkter til ændring af anatomien, fx implantater med kosmetisk formål
- Produkter til injektion, f.eks. fillers i ansigtet
- Produkter til at fjerne fedtvæv, fx udstyr til fedtsugning
- Produkter med elektromagnetisk stråling, fx laser til fjernelse af tatoveringer eller hår
- Produkter til hjernestimulering med strøm eller elektromagnetiske felter, f.eks. til at stimulere hukommelse eller koncentration
Reglerne træder i kraft den 22. juni 2023
Reglerne for produkter uden medicinsk formål træder i kraft den 22. juni 2023, og produkterne skal opfylde kravene i forordning 2022/2346.
Produkterne skal klassificeres efter klassifikationsreglerne i forordningen for medicinsk udstyr, men der gælder særlige klassifikationsregler for de produkter uden medicinsk formål, der er aktive (elektriske). De regler finder man i forordning 2022/2347.
CE-mærkning
Produkter uden et medicinsk formål skal ikke godkendes af Lægemiddelstyrelsen, men produktet skal – som udgangspunkt – CE-mærkes i overensstemmelse med forordningen for medicinsk udstyr (MDR), før det må markedsføres.
CE-mærkning er dokumentationen for, at produktet opfylder den gældende EU-lovgivning (MDR). Formålet med CE-mærkningen er, at produkter frit kan markedsføres i EU, forudsat at produktet opfylder nationale krav (f.eks. det danske krav om registrering og sprogkrav).
Det er fabrikanten, som er ansvarlig for at produktet CE-mærkes, og som en del af det skal fabrikanten klassificere produktet.
Overordnet findes der 4 risikoklasser:
- klasse I
- Klasse IIa
- Klasse IIb
- Klasse III
Klasse I er forbundet med den laveste risiko, mens klasse III er forbundet med den højeste risiko.
Ved CE-mærkning af produkter i klasse I er fabrikanten ansvarlig for processen, mens fabrikanten skal benytte et bemyndiget organ ved klasse IIa, IIb samt III. Bemyndigede organer skal sikre, at fabrikanter af produkter har den påkrævede tekniske dokumentation og kvalitetskontrol for processer. Fabrikanter kan vælge, hvilket bemyndiget organ de vil benytte.
Aktørroller og deres forpligtelser
De forskellige aktørroller indenfor produkter uden et medicinsk formål er defineret således:
Fabrikant
En virksomhed eller person, som fremstiller eller nyistandsætter produkter uden et medicinsk formål, eller som får produkter designet, fremstillet eller nyistandsat og markedsfører det pågældende produkt i eget navn eller under eget varemærke.
System- eller behandlingspakkefabrikant
En virksomhed eller person, som fremstiller eller nyistandsætter system- eller behandlingspakker, eller som får sådanne pakke designet, fremstillet eller nyistandsat og markedsfører den pågældende pakke i eget navn eller under eget varemærke.
Autoriseret repræsentant
En virksomhed eller en person, som er stedfortræder for en fabrikant lokaliseret udenfor EU/EØS.
Importør
Virksomhed eller person, som bringer et produkt fra et land uden for EU/EØS i omsætning på EU-markedet.
Distributør
Virksomhed eller person, som gør et produkt tilgængeligt på det danske marked indtil ibrugtagning.
Specialforretning
Forretning eller person, der er specialiseret i salg af produkter uden et medicinsk formål i risikoklasse IIa, IIb, III og aktivt implantabelt produkter. Minimum 50 % af forretningens varesortiment og omsætning skal udgøres af de nævnte typer produkter for, at forretningen betragtes som en specialforretning.
Hvis aktørerne er etableret i Danmark, er de underlagt europæisk og dansk lovgivning. Aktørernes forpligtigelser fremgår af lovgivningen og er afhængige af den pågældende aktørrolle. Nogle virksomheder vil have flere aktørroller og derfor forpligtigelser, som går på tværs af rollerne.
Lægemiddelstyrelsen har produceret videomateriale med gennemgang af de mest gængse aktørroller.
Registrering - et lovkrav fra den 22. juni 2023
Fra den 22. juni 2023 er det et lovkrav, at følgende dansketablerede aktører skal være registreret hos Lægemiddelstyrelsen for at kunne markedsføre produkter uden et medicinsk formål:
- fabrikanter
- system- eller behandlingspakkeproducenter
- autoriserede repræsentanter
- importører
- distributører
- specialforretninger af produkter uden et medicinsk formål
Fabrikanter, system- eller behandlingspakkeproducenter, autoriserede repræsentanter og importører kan endvidere vælge at lade sig registrere i Den Europæiske Database for Medicinsk Udstyr (Eudamed). Registrering i Eudamed vil blive obligatorisk, når Eudamed er fuldt funktionsdygtigt (forventeligt i andet kvartal 2026).
Registreringen bidrager til Lægemiddelstyrelsens vejledning af virksomheder, markedsovervågning og behandling af hændelser, og dermed medvirker til at fremme patientsikkerheden.
Man kan registrere sig hos Lægemiddelstyrelsen ved at benytte den nedenstående e-blanket.
Link
Blanket til at registrere virksomhed hos Lægemiddelstyrelsen
(e-blanket, åbner i nyt vindue)
Du modtager en kvittering, når vi har behandlet registreringen. Der kan gå op til 14 dage.
Hvis man vil registrere sig i Eudamen forudsætter det, at man først er registreret i Lægemiddelstyrelsens database. Registrering i Eudamed kan dernæst foretages via en registreringsblanket på Europa-Kommissionens hjemmeside.
Lægemiddelstyrelsen har udarbejdet en vejledning til registrering i Eudamed.
Foruden at foretage en virksomhedsregistrering skal virksomheder, der fremstiller, importerer eller distribuerer produkter uden et medicinsk formål, udover at registrere sig, også indberette til Lægemiddelstyrelsen, hvis de har tilknyttet en sundhedsperson til virksomheden eller yder økonomisk støtte til en sundheds- eller anden fagperson, jf. § 5 b, stk. 5 i lov om medicinsk udstyr.
Gebyrer
Ifølge gældende national lov skal alle aktører betale registreringsgebyr samt årsgebyr til Lægemiddelstyrelsen, afhængigt af hvor mange bekendtgørelser aktøren er omfattet af.
Registreringsgebyret pålægges i forbindelse med en ny registrering.
Lægemiddelstyrelsen har myndighedsansvaret for tilsyn og kontrol af bl.a. fabrikanter, autoriserede repræsentanter, importører og distributører af produkter uden et medicinsk formål.
Årsgebyret fastsættes på baggrund af virksomhedens rolle, produktets risikoklasse og antal ansatte i den registrerede virksomhed. Dette gebyr bliver opkrævet både hos fabrikanter, EU-repræsentanter samt importører og distributører af produkter uden et medicinsk formål.
Gebyrsatserne bliver reguleret årligt og offentliggjort på vores hjemmeside pr. 1. januar.
De gebyrer som Lægemiddelstyrelsen opkræver, sker i henhold til disse bekendtgørelser:
Markedsovervågning og indberetning af hændelser
Fabrikanter af produkter uden et medicinsk formål skal have et markedsovervågningssystem. Et markedsovervågningssystem, også kaldet et post-market surveillance system, er et værktøj, som er en del af kvalitetsstyringssystemet, der tages i brug efter, at udstyret er kommet på markedet.
Markedsovervågningssystemet skal sikre, at oplysninger om ydeevne og sikkerhed ved udstyret bliver modtaget og behandlet systematisk efter markedsføringen. På den måde kan fabrikanten af medicinsk udstyr løbende sikre, at deres produkt opfylder de generelle krav til sikkerhed og ydeevne. Det er nødvendigt med en løbende overvågning af udstyret efter, at det er markedsført og taget i brug, for hele tiden at kunne forbedre produktsikkerheden.
Oplysningerne skal bruges til at forbedre udstyrets sikkerhed og ydeevne, samt at minimere risikoen for gentagelser af ulykker med udstyret – kaldet hændelser.
Overvågningssystemet skal bl.a. også sikre, at alvorlige hændelser og sikkerhedsrelaterede korrigerende handlinger bliver indberettet til myndighederne. Fabrikanten har også pligt til at udføre produktændringer for at minimere eventuelle risici ved produktet.
Eventuelle risici skal inkluderes i fabrikantens risikoanalyse.
Indberetning af hændelser
Fabrikanter samt driftsansvarlige for offentlige og private sygehuse har pligt til at underrette myndighederne om alvorlige hændelser med medicinsk udstyr. Alvorlige hændelser som sker i Danmark skal indberettes til Lægemiddelstyrelsen. Også autoriserede sundhedspersoner, der udøver selvstændig virksomhed uden for sygehusvæsenet, er forpligtede til at indberette alvorlige og formodede alvorlige hændelser til Lægemiddelstyrelsen. Lægemiddelstyrelsen sikrer, at oplysninger om alvorlige hændelser med medicinsk udstyr bliver modtaget og undersøgt.
Alvorlige hændelser kan også indberettes af:
Enhver anden borger (brugere, patienter, pårørende m.fl.) kan indberette alvorlige og formodede alvorlige hændelser til Lægemiddelstyrelsen. Når Lægemiddelstyrelsen modtager indberetningen, bliver oplysningerne sendt til fabrikanten, som herefter også indsender en indberetning og undersøger den alvorlige hændelse.
Alvorlige hændelser omfatter:
- en patients, brugers eller anden persons dødsfald
- midlertidig eller varig alvorlig forringelse af en patients, brugers eller anden persons sundhedstilstand
- en alvorlig trussel mod folkesundheden
Hændelser, der kunne have ført til en alvorlig hændelse, men hvor hændelsen blev afværget pga. indgreb fra sundhedspersonale, skal også indberettes.
Sikkerhedskorrigerende handlinger
Sikkerhedsrelaterede korrigerende handlinger, også kaldet Field Safety Corrective Actions (FSCA), er korrigerende handlinger foretaget af en fabrikant for at forebygge eller mindske risikoen for en alvorlig hændelse ved, der er gjort tilgængeligt på markedet.
Fabrikanten er også forpligtet til at iværksætte eventuelle nødvendige korrigerende handlinger og straks informere Lægemiddelstyrelsen.
Fabrikanten skal underrette Lægemiddelstyrelsen om enhver teknisk eller medicinsk grund, som har ført til, at fabrikanten systematisk har trukket udstyr, som tilhører samme type, tilbage fra markedet.
Importører og distributører af medicinsk udstyr og produkter uden medicinsk formål
Både importører og distributører skal indgå i markedsovervågningen af det udstyr, de hhv. bringer i omsætning (importører) eller gør tilgængeligt (distributører).
Importøren skal føre et register over:
- alle klager
- ikke-overensstemmende udstyr
- tilbagetrækninger og tilbagekaldelser
og give fabrikanten, den autoriserede repræsentant og distributørerne de oplysninger, de beder om, så de kan undersøge eventuelle klager.
Hvis en importør eller en distributør opdager eller har mistanke om, at udstyret ikke lever op til kravene i forordningen, må importøren ikke bringe udstyret i omsætning og skal underrette fabrikanten og den autoriserede repræsentant om det, de har opdaget eller har mistanke om. Hvis importøren mener, at udstyret udgør en alvorlig risiko eller er forfalsket, skal importøren også give besked til Lægemiddelstyrelsen.
Hvis en importør modtager en klage eller en indberetning fra en sundhedsperson, skal importøren straks sende klagen eller indberetningen til fabrikanten eller den autoriserede repræsentant.
Distributører og paralleldistributører skal på samme vis sende en klage eller indberetning, som de har modtaget fra sundhedspersoner, patienter eller brugere, til fabrikanten eller den autoriserede repræsentant og importøren.
Læs mere
Vejledning til importører og distributører af medicinsk udstyr (Retsinformation)
Vejledning til nystartede fabrikanter af medicinsk udstyr (Retsinformation) og
Vejledning til fabrikanter af medicinsk udstyr om markedsovervågning (Retsinformation)
Overgangsbestemmelser for produkter uden medicinsk formål
Da de nye regler kun omfatter nye produkter på markedet, er der vedtaget overgangsordninger, som gør, at fabrikanter af denne type udstyr, får tid til at opfylde de nye krav.
Klik på nedenstående indholdsfortegnelse for at læse om overgangsbestemmelserne for produkter uden medicinsk formål:
- Overgangsperiode for produkter uden et medicinsk formål, hvor fabrikanten skal lave en klinisk afprøvning, og hvor et bemyndiget organ er involveret i overensstemmelsesvurderingen
- Overgangsperiode for produkter uden et medicinsk formål, hvor fabrikanten ikke skal lave en klinisk afprøvning, men hvor et bemyndiget organ er involveret i overensstemmelsesvurderingen
- Overgangsperiode for produkter uden et medicinsk formål, som er certificeret af EU’s direktiv om medicinsk udstyr, hvor fabrikanten ikke skal lave en klinisk afprøvning, men hvor et bemyndiget organ er involveret i overensstemmelsesvurderingen
1. Overgangsperiode for produkter uden et medicinsk formål, hvor fabrikanten skal lave en klinisk afprøvning, og hvor et bemyndiget organ er involveret i overensstemmelsesvurderingen
Læs her hvad et bemyndiget organ er: Bemyndigede organer (laegemiddelstyrelsen.dk)
Overgangsperioden starter den 22. juni 2023 og slutter den 31. december 2029. I løbet af denne periode skal forskellige betingelser opfyldes. Nogle gælder for hele perioden, mens andre gælder på bestemte datoer. Nedenfor kan du se, hvilke betingelser der skal være opfyldt, for at produktet kan være på markedet under overgangsperioden.

Figur 1: Betingelser for anvendelse af overgangsperioden i medfør af artikel 2, stk. 1 i EU-Kommissionens fælles specifikationer
(Download figur 1 som pdf)
|
Fra den 22. juni 2023 til den 31. december 2029 |
||
|
||
|
Fra den 23. juni 2024 |
Fra den 23. december 2024 |
Fra den 1. januar 2028 |
| Sponsor (en person, firma, institution eller organisation) har modtaget en bekræftelse på, at ansøgningen om den kliniske afprøvning af produktet er afsluttet, og at den kliniske afprøvning falder ind under anvendelsesområdet for regulativerne om medicinsk udstyr (artikel 2, stk.1, andet afsnit). | Sponsor (en person, firma, institution eller organisation) har indledt den kliniske afprøvning (artikel 2, stk.1, tredje afsnit). | Det bemyndigede organ og fabrikanten har underskrevet en skriftlig aftale om udførelse af overensstemmelsesvurderingen (artikel 2, stk.1, fjerde afsnit). |
Tabel 1: Betingelser for anvendelse af overgangsperiode i medfør af artikel 2, stk. 1 i EU-Kommissionens fælles specifikationer.
Bemærk, at produkter uden et medicinsk formål, som er certificeret af EU’s direktiver om medicinsk udstyr (Medical Device Directives), ikke kan anvende denne overgangsperiode. Se tabel 3.
Læs mere om direktiverne om medicinsk udstyr: EU-Direktiver om medicinsk udstyr (laegemiddelstyrelsen.dk)
2. Overgangsperiode for produkter uden et medicinsk formål, hvor fabrikanten ikke skal lave en klinisk afprøvning, men hvor et bemyndiget organ er involveret i overensstemmelsesvurderingen
Læs her hvad et bemyndiget organ er: Bemyndigede organer (laegemiddelstyrelsen.dk)
Overgangsperioden starter den 22. juni 2023 og slutter den 31. december 2028. I løbet af denne periode skal forskellige betingelser opfyldes. Nogle gælder for hele perioden, mens andre gælder på bestemte datoer. Nedenfor kan du se, hvilke betingelser der skal være opfyldt, for at produktet kan være på markedet under overgangsperioden.
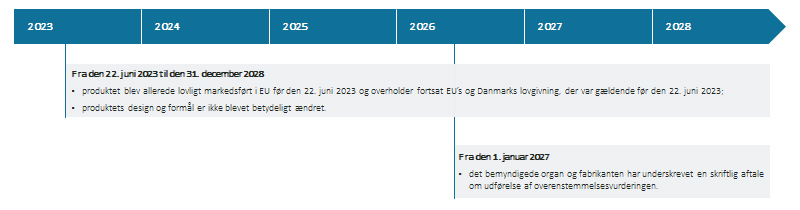
Figur 2: Betingelser for anvendelse af overgangsperioden i medfør af artikel 2, stk. 2 i EU-Kommissionens fælles specifikationer
(Download figur 2 som pdf)
|
Fra den 22. juni 2023 til den 31. december 2028 |
|
|
Fra den 1. januar 2027 |
|
Tabel 2: Betingelser for anvendelse af overgangsperioden i medfør af artikel 2, stk. 2. i EU-Kommissionens fælles specifikationer
Bemærk også her, at produkter uden et medicinsk formål, som er certificeret af EU’s direktiver om medicinsk udstyr (Medical Device Directives), ikke kan anvende denne overgangsperiode. Se tabel 3.
Læs mere om direktiverne om medicinsk udstyr: EU-Direktiver om medicinsk udstyr (laegemiddelstyrelsen.dk)
3. Overgangsperiode for produkter uden et medicinsk formål, som er certificeret af EU’s direktiv om medicinsk udstyr, hvor fabrikanten ikke skal lave en klinisk afprøvning, men hvor et bemyndiget organ er involveret i overensstemmelsesvurderingen
Læs her hvad et bemyndiget organ er: Bemyndigede organer (laegemiddelstyrelsen.dk)
Overgangsperioden slutter den 31. december 2027 for højere risiko-produkter. Disse produkter er alt udstyr i klasse III og for implantabelt udstyr i klasse IIb, bortset fra suturer, hæfteklemmer, tandfyldninger, tandbøjler, tandkroner, skruer, kiler, plader, tråd, nåle, klemmer og forbindelsesled (MDR, artikel 120, stk. 3a, litra a) og den 31. december 2028 for lavere risiko-produkter, hvilket er alle andre klasser og produkter undtaget fra den højere risikoklasse (MDR, artikel 120, stk. 3a, litra b).
Ikke alle produkter, som er certificeret af EU’s direktiver om medicinsk udstyr, kan anvende overgangsperioden fastlagt i artikel 2, stk. 3 i EU-Kommissionens fællesspecifikationer. Denne overgangsperiode er kun gældende, når et certifikat fra EU’s direktiver om medicinsk udstyr blev udstedt efter den 25. maj 2017, stadig var gyldigt den 26. maj 2021, ikke tilbagekaldt efterfølgende, udløb før den 20. marts 2023 og opfylder følgende betingelser oplistet i artikel 120, stk. 2, 2. afsnit, litra a) og b):
- fabrikanten og et bemyndiget organ har ikke inden certifikatets udløbsdato indgået en skriftlig aftale om overensstemmelsesvurdering for så vidt angår udstyr, der er omfattet af det udløbne certifikat, eller udstyr, der er beregnet til at erstatte udstyret.
- Lægemiddelstyrelsen har ikke givet dispensation i medfør af artikel 59, stk. 1 eller artikel 97, stk. 1 i EU’s afløser for direktiverne om medicinsk udstyr, regulativerne om medicinsk udstyr (Medical Device Regulations).
Læs mere om regulativerne om medicinsk udstyr: EU-regler om medicinsk udstyr (laegemiddelstyrelsen.dk)
I løbet af overgangsperioden skal yderligere betingelser opfyldes. Nogle gælder for hele perioden, mens andre gælder på bestemte datoer. Nedenfor kan du se, hvilke betingelser der skal være opfyldt, for at produktet kan være på markedet under overgangsperioden.
|
Fra den 22. juni 2023 til den 31. december 2027 for højere risiko-produkter eller til den 31. december 2028 for lavere risiko-produkter |
|
|
|
|
Fra den 26. maj 2024 |
Fra den 26. september 2024 |
|
|
Tabel 3: Betingelser for anvendelse af overgangsperioden i medfør af artikel 2, stk. 3 i EU-Kommissionens fælles specifikationer
Hvis ovenstående betingelser i artikel 120, stk. 3c, 3d og 3e er opfyldt i regulativerne om medicinsk udstyr, kan disse produkter markedsføres eller tages i brug indtil datoerne fastsat i artikel 120, stk. 3a, litra a og b i regulativerne om medicinsk udstyr, også efter certifikatets udløb.
