
Medicinsk udstyr
Medicinsk udstyr er produkter, der bruges til at diagnosticere, forebygge, lindre eller behandle sygdomme, handicap eller skader. Der findes mere end 500.000 forskellige typer medicinsk udstyr, som kan være alt fra kørestole og briller til diagnostiske analyser, pacemakere, apps på mobiltelefoner og avanceret operationsudstyr.
Medicinsk udstyr er reguleret af to EU-forordninger, hvis overordnede formål er at sikre patientsikkerheden. Forordningerne angiver bl.a. at fabrikanten skal have dokumentation for at udstyret virker efter hensigten og at fordele ved brugen opvejer eventuelle risici. Fabrikanter skal desuden overvåge udstyrets sikkerhed og ydeevne efter udstyret er sat på markedet.
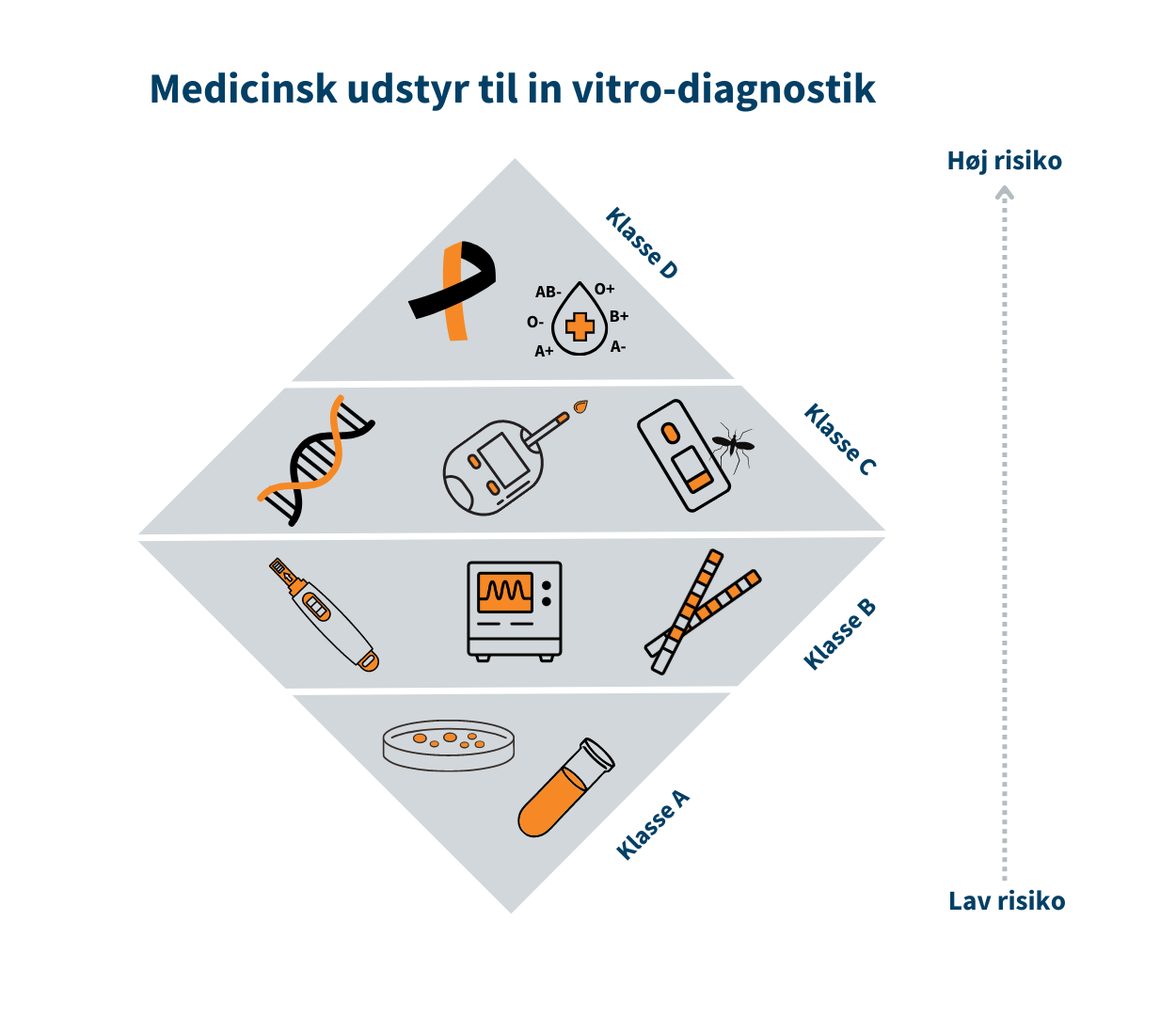
De to forordninger på området er hhv. EU-forordning om medicinsk udstyr og EU-forordning om medicinsk udstyr til in vitro-diagnostik (IVD). Principperne i de to forordninger er ens, men de vedrører to overordnede typer af medicinsk udstyr. IVD er en særlig type medicinsk udstyr som er er bestemt til at undersøge prøvemateriale fra det menneskelige legeme. IVD kan f.eks. være COVID-selvtest, graviditetstest og avancerede laboratorieanalyser på blodprøver til diagnostik af diverse sygdomme og handicap.

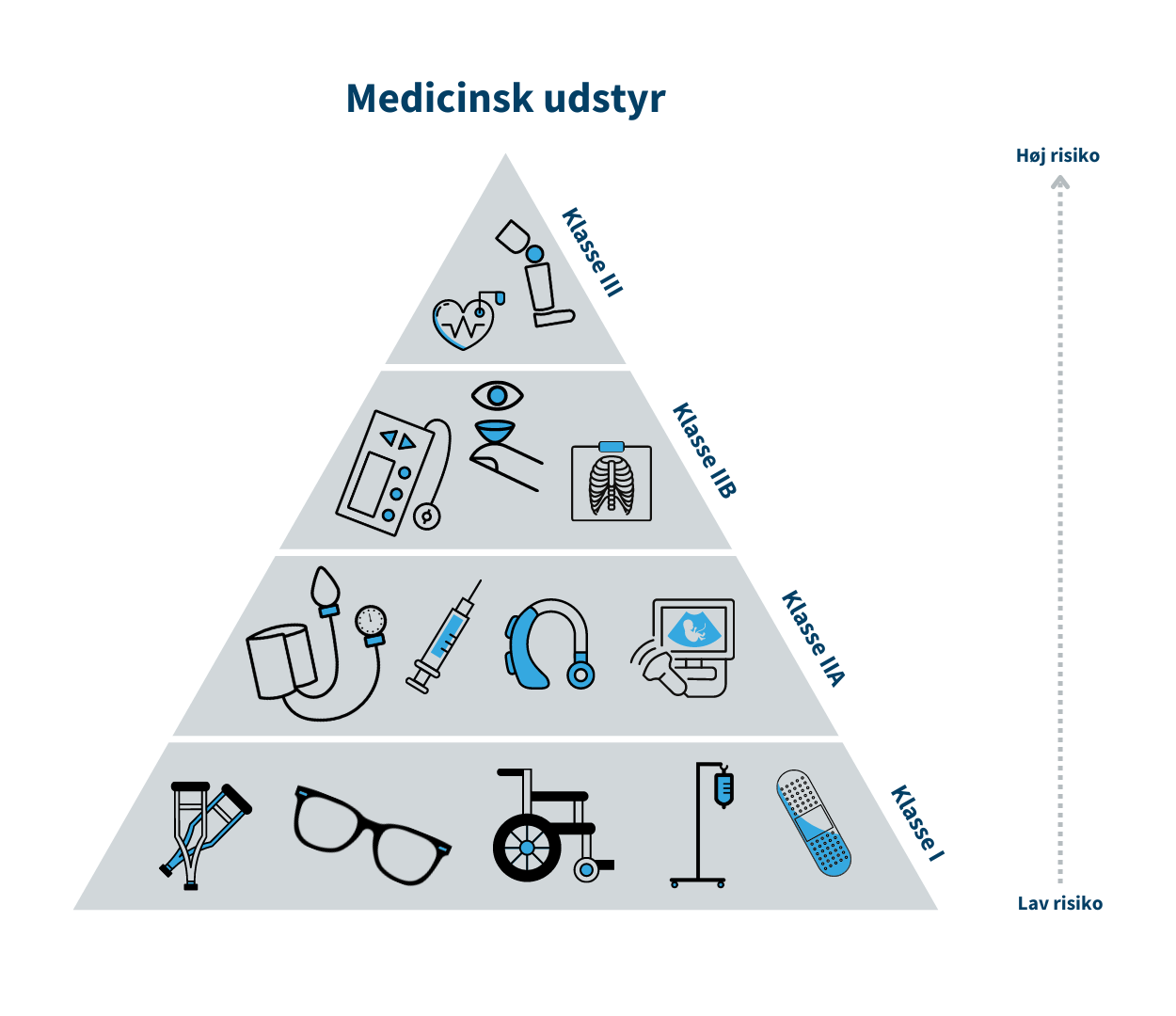
Medicinsk udstyr er inddelt i risikoklasser, som bl.a. skal bruges til at anvise hvilke krav til sikkerhed og ydeevne, der skal være opfyldt før fabrikanten kan markedsføre udstyret i Danmark.Klasse I/Klasse A er forbundet med den laveste risiko, mens klasse III/Klasse D er forbundet med den højeste risiko. Klasse I er eksempelvis en rollator, mens brystimplantater er klasse III. Tilsvarende for medicinsk udstyr til in vitro-diagnostik (IVD) er klasse A eksempelvis en prøvebeholder, mens HIV-test er klasse D.
CE-mærkning for sikkerhed og ydeevne
Medicinsk udstyr skal være CE-mærket, før det kommer på markedet. CE-mærkningen viser, at udstyret opfylder den gældende EU-lovgivning. Hvis der er tale om medicinsk udstyr i en risikoklasse, der er højere end klasse I/A, skal vurderingen af, om produktet kan CE-mærkes, foretages af et såkaldt bemyndiget organ. Et bemyndiget organ er en privat virksomhed, som har fået autorisation til at vurdere den dokumentation, som den virksomhed, der ønsker at sælge produktet, fremlægger. Det bemyndigede organ vurderer, om dokumentationen for produktets sikkerhed og ydeevne er tilstrækkelig til, at udstyret kan CE-mærkes.
Lægemiddelstyrelsen godkender ikke medicinsk udstyr. Lægemiddelstyrelsens opgaver er bl.a. at føre tilsyn med danske fabrikanter og andre aktører indenfor medicinsk udstyr. Lægemiddelstyrelsen reagerer ved kendskab til fejl, svigt eller mangler ved medicinsk udstyr. Dette gøres bl.a. på baggrund af relevante indberetninger om hændelser med medicinsk udstyr, som modtages fra virksomheder, sundhedsprofessionelle eller borgere.
Indberetninger af fejl, svigt og mangler
Lægemiddelstyrelsen modtager indberetninger om fejl, svigt og mangler med medicinsk udstyr. Hændelsesindberetningerne er en vigtig brik i overvågningen af sikkerheden ved medicinsk udstyr. I Lægemiddelstyrelsen bruges indberetningerne i arbejdet med overvågning af medicinsk udstyr i Danmark. Indberetningerne stammer fra sundhedspersonale, fabrikanter, distributører og importører af medicinsk udstyr. Borgere kan også indberette hændelser.