Klassificering af medicinsk udstyr til in vitro-diagnostik (IVD)
Opdateret 1. november 2022
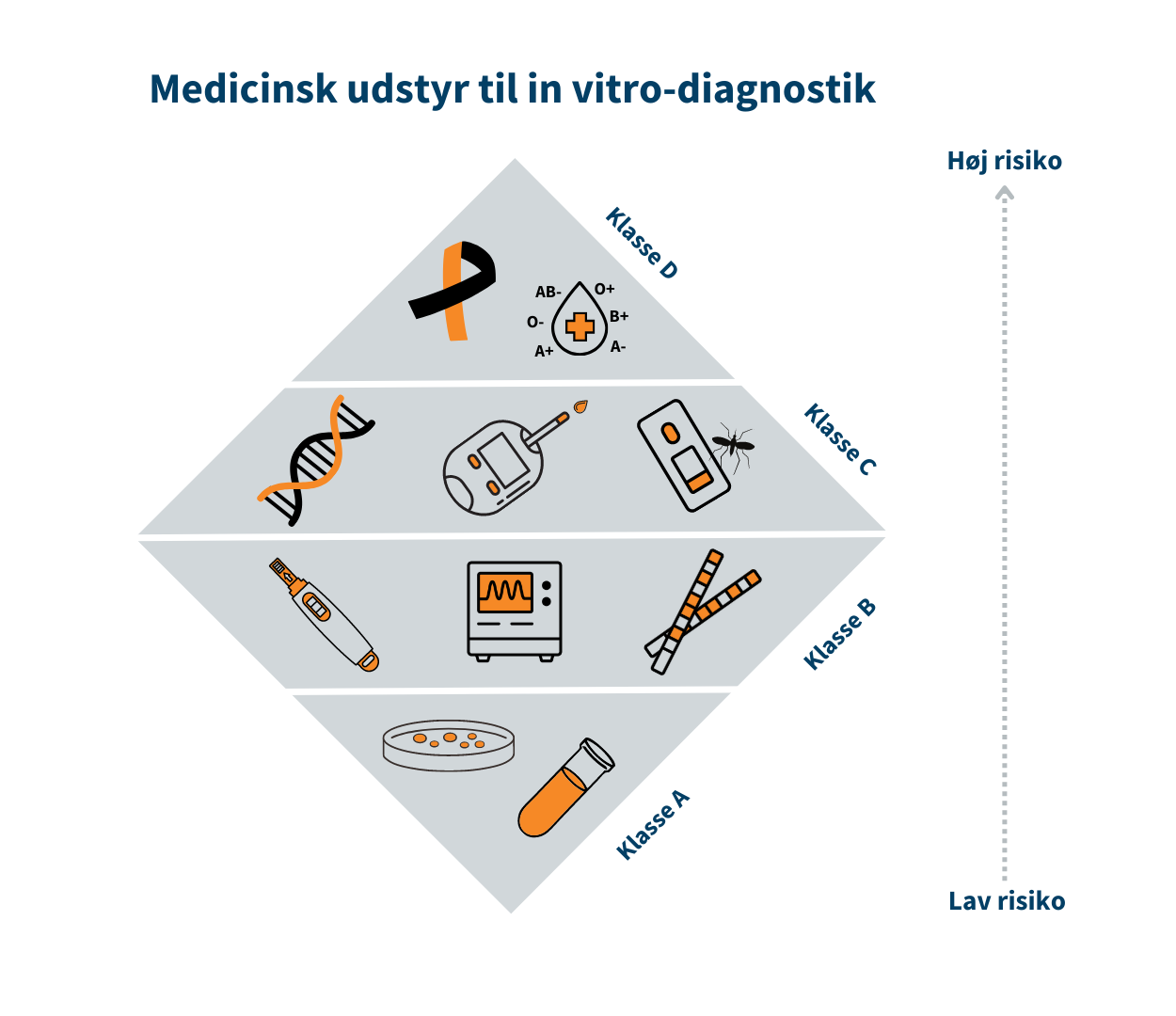
Medicinsk udstyr til in vitro-diagnostik inddeles i risikoklasser som anvises i EU-forordning 2017/746 om medicinsk udstyr til in vitro-diagnostik bilag VIII.
IVD, som af fabrikanten er beregnet til selvtestning, skal følge en overensstemmelsesvurderingsprocedure med involvering af et bemyndiget organ, som skal udstede et certifikat til fabrikanten før udstyret retmæssigt kan CE-mærkes og markedsføres i EU. Desuden er der et krav i dansk lovgivning om at medicinsk udstyrs mærkning og brugsanvisning skal være på dansk samt at brugsanvisningen skal ledsage eller emballeres sammen med et eller flere eksemplarer af udstyret.