
Vejledning målrettet ikke-kommercielle kliniske forsøg
At være sponsor i et klinisk forsøg medfører ansvar og forpligtigelser, som defineret i ICH-GCP E6(R3).
Som sponsor er det essentielt at have et overblik over de regulatoriske krav samt de processer og arbejdsgange, der er forbundet med disse krav.
Klik på den interaktive vejviser herunder og få overblik over regelsættene.
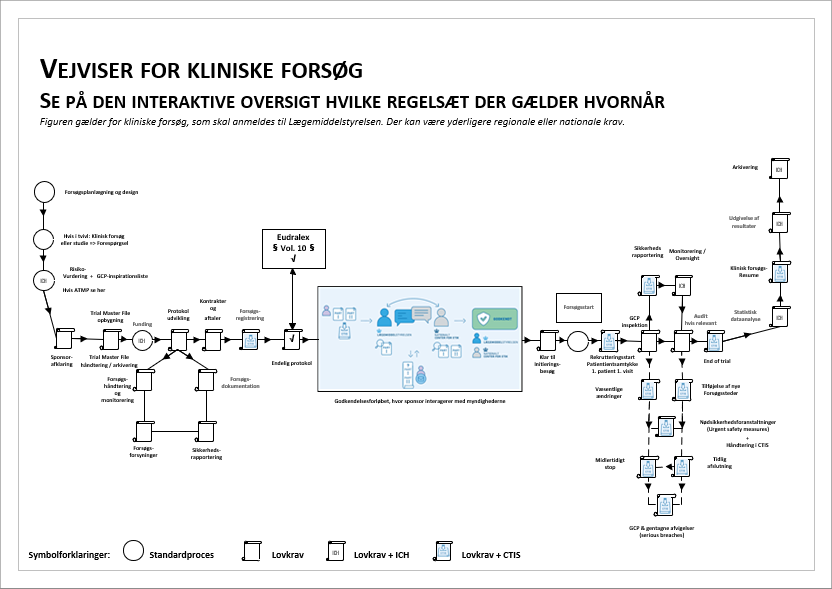
Med ikrafttrædelsen af forordning 536/2014 for kliniske forsøg har myndighederne i Europa fået arbejdsbetingelser, som sikrer en harmoniseret godkendelsesproces, samtidigt med at de vedtagne tidsfrister skal overholdes. For at imødekomme disse frister og krav kræves det, at sponsorer er grundigt forberedte, når de anmelder det kliniske forsøg, og at kvalitet er integreret i både design og arbejdsgange.
Som myndighed er det vores erfaring, at det kan være en udfordring for ikke-kommercielle sponsorer, der ikke nødvendigvis har regulatorisk erfaring, at få et klart overblik over myndighedernes krav og forventninger og dermed leve op til disse.
Denne side er oprettet for at sætte fokus på de emner, som kan være særligt relevante eller udfordrende for non-kommercielle sponsorer. Vi vil med siden også gerne pege på, hvor der er hjælp at hente. Som sponsor er I altid velkomne til at kontakte sektion for kliniske forsøg for yderligere information eller vejledning.
Defintioner på kliniske forsøg
Lav-interventionsforsøg og forsøg med pragmatiske elementer
Regulering af lav-interventionsforsøg
Der er ikke fordele for sponsor ved et lav-interventionsforsøg i forhold til et almindeligt klinisk forsøg.
Vi ser en opfattelse af, at muligheden for udeladelse af individuelt medicinregnskab og risikotilpasset bivirkningshåndtering er tilknyttet lav-interventionsforsøg, men dette er ikke tilfældet.
Begge disse muligheder er til stede i almindelige kliniske forsøg, hvis risikovurdering og retfærdiggørelse er beskrevet i protokollen. Sagsbehandlingstiden er heller ikke anderledes for lav-interventionsforsøg.
Low intervention trial (LIT) er en regulatorisk betegnelse og er ikke det samme som, at man i klinikken betragter forsøget som lav-intervention.
Til udarbejdelse af protokollen vil vi henvise til Risk proportinate approaches in Clinical Trials samt Lægemiddelstyrelsens vejledning om Risikobaseret bivirkningshåndtering.
Kriterier for lav-interventionsforsøg
Som udgangspunkt indbefatter definitionen på lav-interventionsforsøg, at forsøgslægemidlet er markedsført, bruges i overensstemmelse med produktresume (SmPC), samt ikke påfører forsøgspersoner mere end minimal byrde.
Følgende tages i betragtning ved den forsøgsspecifikke retfærdiggørelse af lav-interventionsforsøg:
- Retfærdiggørelsen er ikke en klinikers ”almindelige” risikovurdering, men sponsors retfærdiggørelse, som skal være baseret på alle punkterne i lovgivningen (se nedenfor)
- Henvisninger til at andre forsøg, der er klassificeret som lav-intervention, kan ikke bruges da vurderingen for LIT-status er forsøgsspecifik
- Der opfordres til at lave en solid retfærdiggørelse ved indsendelse af ansøgningen. Med mindre man følger behandlingsvejledningen i produktresume (SmPC) fuldstændigt eller har en specifik klinisk-guideline (national eller europæisk) at henvise til, opfordres sponsor til at lave en detaljeret retfærdiggørelse for LIT-status, baseret på litteraturgennemgang for solid evidens, afdelingens erfaring (inkl. data for både antal behandlede patienter og antal år), samt en grundig sikkerheds vurdering af afdelingens erfaring og en konklusion på at evidensen er på linje med kliniske guidelines
- En LIT status vurderes både ”klinisk” og etisk, så aspekter som kan tilføje ekstra gener for forsøgsdeltageren, så som ekstra blodprøver/biopsier osv. skal retfærdiggøres både fra et klinisk og etisk perspektiv.
Definition på lav-interventionsforsøg i Klinisk forsøg forordningen:
»Klinisk lav-interventionsforsøg«: et klinisk forsøg, som opfylder alle nedenstående betingelser:
a) forsøgslægemidlerne, bortset fra placebo, er godkendt
b) ifølge protokollen for det kliniske forsøg
i) anvendes forsøgslægemidlerne i overensstemmelse med vilkårene i markedsføringstilladelsen, eller
ii) anvendelsen af forsøgslægemidler er evidensbaseret og understøttet af offentliggjort videnskabelig evidens for sikkerheden og effekten af disse forsøgslægemidler i en af de berørte medlemsstater
c) de ekstra diagnose- eller kontrolprocedurer udgør kun en minimal ekstra risiko eller byrde for forsøgspersonernes sikkerhed sammenlignet med normal klinisk praksis i en berørt medlemsstat.
Hvad er et pragmatisk forsøg?
Der findes ingen definiton på et pragmatisk forsøg i lovgivningen i EU. ICH GCP E6 (R3) beskriver at forsøg kan have pragmatiske elementer: “Pragmatic elements in clinical trials are those that integrate aspects of clinical practice into the design and conduct of the trial (e.g., simplified protocols with streamlined data collection)”.
At kalde et forsøg pragmatisk giver ingen særlige regulatoriske fordele.
Til udarbejdelse af protokollen vil vi henvise til Risk proportinate approaches in Clinical Trials samt Lægemiddelstyrelsens vejledning om Risikobaseret bivirkningshåndtering.
.
Sponsors rolle i - og ansvar for kliniske forsøg
Et klinisk forsøg skal have en sponsor, der påtager sig det overordnede ansvar for forsøget.
Det skal i hvert enkelt forsøg entydigt fremgå, hvem der har påtaget sig dette sponsoransvar.
Det gælder ikke mindst, når et hospital eller en hospitalsafdeling anmeldes som sponsor for et klinisk lægemiddelforsøg. Forordning (EU) 536/2014 om kliniske forsøg med lægemidler, lov om kliniske forsøg, og guideline for god klinisk praksis (ICH GCP), beskriver disse forpligtelser.
Se nærmere i Q&A-dokumentet, som beskriver Lægemiddelstyrelsens forventninger til sponsors rolle i forhold til organisering og ansvar.
Yderligere information om definitioner på kliniske lægemiddelforsøg
Skabeloner til ansøgning
Kontraktskabelon
Ny protokolskabelon (Oktober 2025)
Vi har lavet en ny protokolskabelon, som indeholder endnu mere vejledning fra både de Videnskabsetiske Medicinske Komiteer og Lægemiddelstyrelsen.
Ved at tage afsæt i denne nye skabelon sikres det, at de regulatoriske krav til protokollen i højere grad overholdes – og forhåbentlig gør protokoludviklingen både nemmere og bedre.
Vi vil opfodre til at anvende denne protokolskabelon (i stedet for en på afdelingen tidligere anvendt skabelon).
Teknisk vejledning (CTIS m.m.)
Generel information
Nedenfor ses de fejl og-/eller mangler der oftest ses i forbindelse med validering af en forsøgsansøgning eller væsentlige ændringer (substantial modification), og derved udløser considerations til sponsor.
Med denne liste håber vi at kunne forebygge fejl og facilitere processen.
Vi gør endvidere opmærksom på, at sponsor ikke opkræves gebyr før indsendelsen er erklæret valid. Der skal derfor ikke betales noget gebyr, hvis det indsendte afvises af myndighederne, eller sponsors frister udløber under denne fase.
Find mere information på vores generelle Spørgsmål og Svar-side vedrørende kliniske forsøg.
Opmærksomhedspunkter fra GCP-inspektørerne vedr. elektroniske systemer
Lægemiddelstyrelsens GCP-inspektører har over en periode gennemført et projekt med fokus på håndtering af systemer og data i investigatorinitierede forsøg. Projektet har dækket mange områder, hvoraf nogle af de vigtigste har været opgaver, der er lagt i kontrakt vedrørende f.eks. levering af systemer til randomisering og dataindsamling, samt dokumentation for processer efter sidste forsøgsperson har forladt forsøget, såsom dataanalyser, afblinding og dokumentation for aktiviteter i Trial Master File.
Projektet blev gennemført såvel for at øge forskernes og regionernes fokus på at få valide konklusioner af forsøgene, som for at vejlede og sætte standarder på svære områder, der ikke tidligere har været så hyppigt inspiceret.
Strukturerede data
1. Opdatering af eksisterende dokumenter til nye versioner:
Til at uploade en ny version af et dokument skal ”Update” knappen (Papir-ikonet) anvendes. Brug ikke ”Add document”, som kun er til nye dokumenter.
2. Versionering og navngivning af dokumenter:
Korrekt navngivning af dokumenter og dato/versionering angives i de strukturerede data felter i CTIS. Undgå at sætte dato/version i selve filnavnene.
Sektionen Form
1. Skabelon til følgebrev
Vi tilråder at sponsor anvender Template til Cover Letter (Fra CTCG); Initial application, Substantial Modification, Modification description
Den seneste udgave, kan findes på CTCG’s hjemmeside under bjælken "Key documents list".
2. Faktureringsoplysninger:
Angiv tydeligt hospitalsafdling, EAN og CVR nummer, + evt. særlige referencer der ønskes medtaget på fakturaen.
Sektionen Part I
Sektionen Part I, Trial details, Protocol information
1. Angivelse af navne i protokollen:
Det bør kun være sponsors navn der er angivet i protokollen. Navne på co-investigatorer, samarbejdspartnere, laboratorier m.m. bør udelades af protokollen, for at undgå unødige substantial modifications, hvis disse ændrer sig.Sektionen Part I, Sponsors
2. Monitor:
Husk at angive monitor i Third parties. Se evt. afsnit 2.6 i GCP enhedernes vejledning.Sektionen Part I, Products
3. Feltet "Excluded MSCs":
Dette felt anvendes kun ved multinationale forsøg, hvor ikke alle lægemidler anvendes i alle lande.4. Upload af produktresumé (SmPC)
Ved forsøg hvor der udelukkende anvendes markedsførte lægemidler indenfor indikation, er det kun nødvendigt at indsende deres respektive produktresumé (SmPC), som kun skal uploades i dets tilhørende fane under Investigator´s Brochure. Det er ikke nødvendigt at uploade det mere en én gang, eller under andre faner, selvom disse faner kan være markeret med et stjernesymbol.Non-substantielle / Ikke væsentlige ændringer og hvordan de uploades i CTIS
Det er nu blevet muligt at indsende ikke-væsentlige ændringer, der førhen skulle indsendes via en substantial modification.
Dette indbefatter blandt andet forlængelse af forsøgsperioden.
Se den fulde liste af nye CTIS funktioner for ikke-væsentlige ændringer længere nede på siden.
Der er to typer ikke-væsentlige ændringer:
- Non-substantielle ændringer (NSM)
- 81.9 non-substantielle (81.9 NSM)
81.9-NSM skal opdateres løbende af sponsor i CTIS i løbet af forsøgsperioden.
Andre NSM’er skal opdateres i CTIS sammen med den næste væsentlige ændring (SM) eller senest ved forsøgets afslutning, hvis der ikke er indsendt nogen SM’er i mellemtiden.
81.9-NSM er ændringer i et klinisk forsøg, som ikke er væsentlige ændringer (SMs), men som ikke desto mindre er relevante for de berørte medlemsstaters tilsyn med de kliniske forsøg, skal løbende opdateres af sponsoren i EU-databasen i overensstemmelse med artikel 81, stk. 9, i forordning (EF) nr. 536/2014. For en liste over ikke-væsentlige ændringer henvises til bilag IV i dette dokument: CLINICAL TRIALS REGULATION (EU) NO 536/2014, VERSION 7.1
Det er altid sponsors ansvar at vurdere, om en ændring er non-substantiel (at den ikke påvirker forsøgets videnskabelige værdi eller deltagernes rettigheder og sikkerhed). Det er vigtigt, at denne vurdering kan begrundes ved monitoreringsbesøg og eventuelle inspektioner.
Alle ikke-væsentlige ændringer, både 81.9-NSM og NSM, kræver ikke vurdering og godkendelse af myndighederne før implementering.
Se den fulde liste af nye CTIS funktioner for ikke-væsentlige ændringer herunder:
Fanen MSCs:
- Trial subject numbers
Fanen Part I:
- Trial information - følgende sektioner er nu redigerbare;
- Medical condition
- Trial duration
- Source of monetary support or material support
- Age range (mindreårige kan ikke tilføjes)
- Clinical trial group (Healthy, patients, vulnerable)
- Protocol
- Alle sektioner er redigerbare, nye dokumenter kan uploades, nuværende dokumenter kan opdateres
- Alle sektioner er redigerbare, nye dokumenter kan uploades, nuværende dokumenter kan opdateres
- SA and PIP
- SA kan tilføjes/opdateres
- PIP kan opdateres, hvis der er en eksisterende, men der kan ikke tilføjes en ny
- Associated CTs
- Associated CTs kan tilføjes/slettes
- Document agreement from another sponsor, kan tilføjes/opdateres
- Sponsor
- Sponsorinformationer kan ændres, så længe det ikke er ORG-ID
- Co-sponsor kan slettes, men ikke tilføjes
- Responsibilities kan ændres
- Contact points kan tilføjes/ændres
Sikkerhedsrapportering
SUSARS = Alvorlige uventede bivirkninger
SUSARs
SUSARs skal jf. den nye EU-forordning indberettes direkte til den europæiske EudraVigilance-database. Læs mere her
Årlig sikkerhedsrapportering
Annual Safety Report (ASR)
Den årlige sikkerhedsrapport skal indsendes via CTIS. Bemærk det er nødvendigt at have den rigtige brugerrolle, for at fanen Annual safety reporting er synlig. Se evt. vores guide.
For mononationale forsøg, der kun foregår i Danmark anvendes GCP-enhedens skabelon for årlige sikkerhedsrapporter, den findes her: Hændelser og bivirkninger vejledninger og skabeloner - GCP-Enhederne
For multinationale forsøg (sites både i Danmark og i udlandet) anvendes ASR skabelon fra Heads of Medicines Agency (HMA, CTCG), den findes her: Heads of Medicines Agencies: Clinical Trials Coordination Group under Key Document List => Clinical Trial Safety, Simplified template of Annual Safety Report
Andre sikkerhedsrapporter
Andre sikkerhedsmæssige indberetninger
Ved behov for at advisere Lægemiddelstyrelsen om andre sikkerhedsmæssige forhold som utilsigtede hændelser (Unexpected events), hastende sikkerhedsforanstaltninger (Urgent safety measures) og alvorlige afvigelser (Serious breaches), skal fanen Notifications anvendes.
Temporary Halt og Early Termination (pEOT)
Midlertidig standsning og afslutning af forsøg før tid, som følge af sikkerhedsmæssige årsager, skal registreres i fanen Notifications.
Ændringer:
9. februar, 2026 - Håndtering af ikke-væsentlige ændringer i CTIS
5. februar 2026 - 81.9 non-substantielle ændringers håndtering i CTIS
4. November, 2025 - Q&A om sponsors rolle og ansvar
Oktober 2025 - Opdateret samarbejdsaftale for LMST og VMK + ny protokolskabelon der reflekterer revideret ICH GCP standard.
11.7.2025 - Oversigt henviser til nyeste CTIS vejledning