
Kliniske forsøg - spørgsmål og svar
På denne side vil vores spørgsmål/svar sektion løbende blive opdateret. I kan følge med i opdateringer i vores ændringslog i bunden af denne hjemmeside.
Spørgsmål/svar vedrørende kliniske lægemiddelforsøg under Forordningen
Definitioner på kliniske lægemiddelforsøg
1. Lægemiddelforsøg eller ej?
Hvilke kliniske forsøg skal Lægemiddelstyrelsen godkende?
Vi skal godkende kliniske forsøg med lægemidler og medicinsk udstyr, der ikke er CE mærket til det formål det afprøves til. (Læs mere om medicinsk udstyr her).
2. Hvad er et klinisk forsøg med et lægemiddel?
Jf. artikel 2 i klinisk forsøg forordningen er definitionerne som følger nedenfor.
Kliniske forsøg og kliniske lav-interventionsforsøg skal godkendes før igangsættelse.
Vær opmærksom på de forskellige betegnelser ”klinisk undersøgelse” og ”klinisk forsøg” afhængig af om det er tale om ansøgningspligt.
1) »Klinisk undersøgelse«: enhver undersøgelse vedrørende mennesker, der har til formål:
a) at afdække eller efterprøve de kliniske, farmakologiske eller andre farmakodynamiske effekter af et eller flere lægemidler
b) at identificere bivirkninger ved et eller flere lægemidler, eller
c) at undersøge absorptionen, distributionen, metabolismen og udskillelsen af et eller flere lægemidler med henblik på at vurdere sikkerheden og/eller effekten af disse lægemidler.
2) »Klinisk forsøg«: en klinisk undersøgelse, som opfylder en af følgende betingelser:
a) forsøgspersonens udpegelse til en bestemt terapeutisk strategi afgøres på forhånd og følger ikke normal klinisk praksis i den berørte medlemsstat
b) beslutningen om at ordinere forsøgslægemidlet træffes sammen med beslutningen om at inddrage forsøgspersonen i den kliniske undersøgelse, eller
c) der anvendes diagnose- eller kontrolprocedurer ud over normal klinisk praksis over for forsøgspersonerne.
3) »Klinisk lav-interventionsforsøg«: et klinisk forsøg, som opfylder alle nedenstående betingelser:
a) forsøgslægemidlerne, bortset fra placebo, er godkendt
b) ifølge protokollen for det kliniske forsøg
- anvendes forsøgslægemidlerne i overensstemmelse med vilkårene i markedsføringstilladelsen, eller
- anvendelsen af forsøgslægemidler er evidensbaseret og understøttet af offentliggjort videnskabelig evidens for sikkerheden og effekten af disse forsøgslægemidler i en af de berørte medlemsstater
c) de ekstra diagnose- eller kontrolprocedurer udgør kun en minimal ekstra risiko eller byrde for forsøgspersonernes sikkerhed sammenlignet med normal klinisk praksis i en berørt medlemsstat.
Se mere om reguleringen af lav-interventionsforsøg under punkt 7.
4) »Ikke-interventionsundersøgelse«: en klinisk undersøgelse, der ikke er et klinisk forsøg.
3. Hvornår kan et lægemiddel defineres som et værktøj ?
Lægemidlet er ikke genstand for undersøgelsen og anvendes som værktøj til at opnå et velkendt fysiologisk respons.
Eksempler:
- Pupiludvidende øjendråber anvendes for at undersøge øjets fysiologi
- Radioaktivt mærket sporstof (lægemiddel) ved en PET-scanning for at få et billede af f.eks. iltoptagelse eller glukoseomsætning i kroppen
- Der afdækkes/ efterprøves IKKE terapeutisk, diagnostisk eller forbyggende effekt eller sikkerhed af lægemidlet.
- Der indsamles IKKE data vedrørende lægemidlets farmakologiske virkninger herunder farmakodynamik og/eller –farmakokinetik.
En klinisk undersøgelse, hvor lægemiddel/lægemidler udelukkende bruges som værktøj, defineres ikke som et klinisk forsøg og skal ikke anmeldes til Lægemiddelstyrelsen.
4. Skal jeg anmelde undersøgelser hvor lægemidler anvendes som værktøj til Lægemiddelstyrelsen?
Nej, denne type undersøgelser er ikke omfattet af ansøgningspligten.
5. Kan jeg få en vurdering af, om Lægemiddelstyrelsen skal give tilladelse til et specifikt forsøg?
Hvis du er i tvivl om, klassifikationen af et forsøg eller en undersøgelse og om det skal anmeldes til Lægemiddelstyrelsen, kan du sende en forespørgsel om ansøgningspligten.
Forespørgslen skal indeholde protokollen for forsøget og data om det præparat, der ønskes undersøgt. Hvis protokollen ikke er færdig, kan vi vurdere det ud fra en synopsis, hvor formål, effektmål og beskrivelse af hvordan de opnås, skal fremgå. Du kan sende din forespørgsel via dette link: Forespørgsel om klinisk forsøgs-klassifikation.
Hvis der er tvivl om, hvorvidt stoffet, der skal afprøves, er et lægemiddel eller ej, kan du hente hjælp her: Afgrænsning mellem lægemidler og andre produkter.
6. Hvad er definitionen på et ikke-kommercielt klinisk forsøg?
Et ikke-kommercielt forsøg er defineret ved, at det helt eller overvejende gennemføres uden indflydelse fra lægemiddelvirksomheder. Det er derved forskerinitieret og ingen lægemiddelvirksomheder har indflydelse på forsøgets design, opstart og gennemførelse samt registrering og offentliggørelse af forsøgsresultater.
7. Regulering af lav-interventionsforsøg
Der er ikke fordele for sponsor ved et lav-interventionsforsøg i forhold til et almindeligt klinisk forsøg.
Vi ser en opfattelse af, at muligheden for udeladelse af individuelt medicinregnskab og risikotilpasset bivirkningshåndtering er tilknyttet lav-interventionsforsøg, men dette er ikke tilfældet.
Begge disse muligheder er til stede i almindelige kliniske forsøg, hvis risikovurdering og retfærdiggørelse er beskrevet i protokollen. Sagsbehandlingstiden er heller ikke anderledes for lav-interventionsforsøg.
Low intervention trial (LIT) er en regulatorisk betegnelse og er ikke det samme som, at man i klinikken betragter forsøget som lav-intervention.
Til udarbejdelse af protokollen vil vi henvise til Risk proportinate approaches in Clinical Trials samt Lægemiddelstyrelsens vejledning om Risikobaseret bivirkningshåndtering.
Kriterier for lav-interventionsforsøg (se også punkt 2.3 ovenfor)
Som udgangspunkt indbefatter definitionen på lav-interventionsforsøg, at forsøgslægemidlet er markedsført, bruges i overensstemmelse med produktresume (SmPC), samt ikke påfører forsøgspersoner mere end minimal byrde.
Følgende tages i betragtning ved den forsøgsspecifikke retfærdiggørelse af lav-interventionsforsøg:
- Retfærdiggørelsen er ikke en klinikers ”almindelige” risikovurdering, men sponsors retfærdiggørelse, som skal være baseret på alle punkterne i lovgivningen (se nedenfor)
- Henvisninger til at andre forsøg, der er klassificeret som lav-intervention, kan ikke bruges da vurderingen for LIT-status er forsøgsspecifik
- Der opfordres til at lave en solid retfærdiggørelse ved indsendelse af ansøgningen. Med mindre man følger behandlingsvejledningen i produktresume (SmPC) fuldstændigt eller har en specifik klinisk-guideline (national eller europæisk) at henvise til, opfordres sponsor til at lave en detaljeret retfærdiggørelse for LIT-status, baseret på litteraturgennemgang for solid evidens, afdelingens erfaring (inkl. data for både antal behandlede patienter og antal år), samt en grundig sikkerheds vurdering af afdelingens erfaring og en konklusion på at evidensen er på linje med kliniske guidelines
- En LIT status vurderes både ”klinisk” og etisk, så aspekter som kan tilføje ekstra gener for forsøgsdeltageren, så som ekstra blodprøver/biopsier osv. skal retfærdiggøres både fra et klinisk og etisk perspektiv.
8. Hvad er et pragmatisk forsøg?
Der findes ingen definition på et pragmatisk forsøg i lovgivningen i EU. ICH GCP E6 (R3) beskriver at forsøg kan have pragmatiske elementer: “Pragmatic elements in clinical trials are those that integrate aspects of clinical practice into the design and conduct of the trial (e.g., simplified protocols with streamlined data collection)”.
At kalde et forsøg pragmatisk giver ingen særlige regulatoriske fordele.
Til udarbejdelse af protokollen vil vi henvise til Risk proportinate approaches in Clinical Trials samt Lægemiddelstyrelsens vejledning om Risikobaseret bivirkningshåndtering.
.
Sponsors rolle i - og ansvar for kliniske forsøg
Et klinisk forsøg skal have en sponsor, der påtager sig det overordnede ansvar for forsøget.
Det skal i hvert enkelt forsøg entydigt fremgå, hvem der har påtaget sig dette sponsoransvar.
Det gælder ikke mindst, når et hospital eller en hospitalsafdeling anmeldes som sponsor for et klinisk lægemiddelforsøg. Forordning (EU) 536/2014 om kliniske forsøg med lægemidler, lov om kliniske forsøg, og guideline for god klinisk praksis (ICH GCP), beskriver disse forpligtelser.
Se nærmere i Q&A-dokumentet, som beskriver Lægemiddelstyrelsens forventninger til sponsors rolle i forhold til organisering og ansvar.
Krav til dokumenter
1. Hvor kan jeg se, hvilke dokumenter der skal indsendes?
I forordningens annex 1 er der listet hvilke dokumenter der skal indsendes.
De Videnskabsetiske Medicinske Komitéer har også lavet en Q&A-side for CTR/CTIS, hvor de svarer på en lang række spørgsmål omkring krav til indsendelse.
2. Er der retningslinjer for navngivning af dokumenter?
Dokumenter, der uploades I CTIS portalen må ikke indeholde dato og versionsnummer i filnavnet.
I stedet henvises til “Instruction Naming Documents”.
3. Hvordan indsender og opdaterer jeg dokumenter i CTIS?
Se også sponsor frequently asked questions (FAQ) publiceret af EMA.
3 a) Indsendelse af dokumenter:
Kan gøres ved enten initialansøgning, substantial modification eller non-substantial modification. Dokumenterne uploades enten under de sektioner de tilhører eller under fanen ”All documents”. Første upload af hver type dokument (Cover Letter, Protokol, m.m.) vil som hovedregel uploades ”for publication”. Disse bliver altså offentligt tilgængelige ved publicering og bør ikke indeholde sensitive oplysninger. Dog vil nogle typer dokumenter altid blive uploadet ”not for publication” – såsom IMPD. Find flere oplysninger om dette i The GCP Units guide to CTIS (overall guideline), som findes på GCP-enhedernes hjemmeside.
Transparens
Hvor meget bliver offentliggjort i EU-portalen og databasen (CTIS)?
I overensstemmelse med de reviderede CTIS transparensregler (Revised CTIS transparency rules (europa.eu), er det kun visse dokumenter, som uploades i EU-databasen, der offentliggøres. Det har blandt andet til formål at beskytte personoplysninger og kommercielt fortrolige oplysninger.
Pr. 18. Juni 2024 er CTIS tilpasset de reviderede CTIS transparensregler.
Vi henviser derfor til ECT EU Guidance. Endvidere til quick guide for users, der giver et hurtigt overblik over hvilke dokumenter, der bliver offentliggjort hvornår.
Generelt skal al inklusion af persondata minimeres og såfremt der inkluderes persondata i dokumentversion ”for publication”, skal disse anonymiseres med få undtagelser, hvor informationen er påkrævet at blive publiceret.
Husk at sørge for at de strukturerede data i CTIS (Titel, sprog, version og dato), stemmer overens med det faktiske indhold af dokumentet. Version og dato bør ikke fremgå af dokumentets titel, men kun i de strukturerede data - se evt. CTCG best practice guide naming of documents.
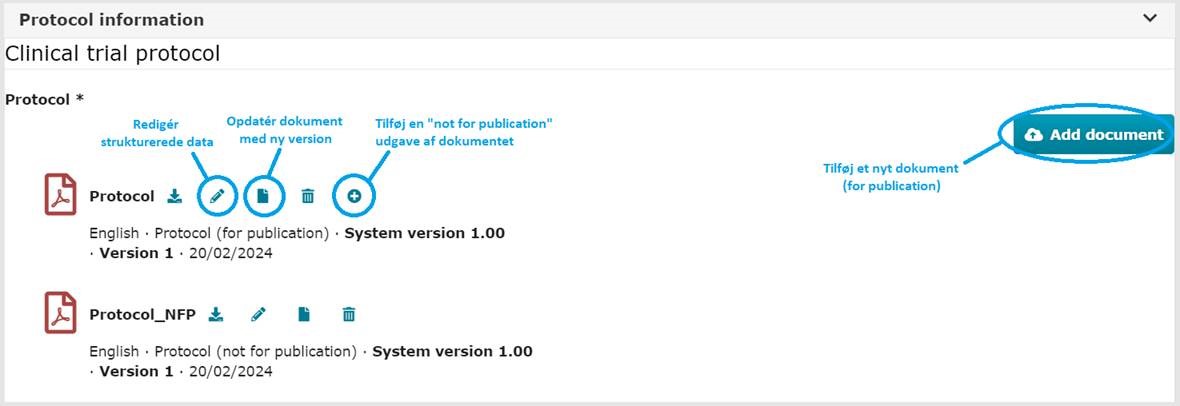
3 b) Opdatering af dokumenter:
Ved upload af nye versioner af eksisterende dokumenter, anvendes ”Update”-knappen (papir ikonet) ud for det eksisterende dokument.
4. Hvilke oplysninger skal forsøgsprotokollen indeholde?
Lægemiddelstyrelsen har lavet en template for forsøgsprotokoller (på engelsk som Word-fil), der beskriver hvilke oplysninger der skal fremgå af en protokol for at overholde kravene i forordningens annex 1, afsnit D samt the International Council for Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) M11 template
Vi har opdateret vores protokolskabelon, så den afspejler kravene i den nyligt reviderede ICH GCP-standard. Blandt andet er begrebet sponsor oversight nu indarbejdet i skabelonen.
Se også ACT EUs vejledning.
5. Hvilke oplysninger skal fremgå af følgebrevet (cover letter) ved indsendelse af ansøgning?
Følgebrevet skal, jf. bilag 1, afsnit B i forordningen, indeholde kerneoplysninger om forsøget såsom EU-forsøgsnummer, protokolkode/nummer, titel og sponsor. Det skal fremgå af følgebrevet, hvem sponsor har uddelegeret GCP-monitoreringen af forsøget til og det skal bekræftes, at monitor forstår dansk. Faktureringsoplysninger kan nævnes i følgebrevet eller uploades som et særskilt dokument i sektionen ”Proof of payment”. Der skal være en liste over alle forsøgslægemidler og hjælpelægemidler i forsøget, med angivelse af deres regulatoriske status (markedsført eller ej). Det skal angives hvor I ansøgningen at referencesikkerhedsdokumenterne (RSI) for de enkelte forsøgslægemidler findes.
Er der nogen særlige omstændigheder ved forsøgets design, forsøgslægemidler eller population, skal det også fremgå af følgebrevet, såsom first-in-human forsøg, decentrale forsøg, forsøg på børn eller særligt sårbare populationer m.fl. For en udførlig liste henviser vi til bilag 1, afsnit B i forordningen, der beskriver krav til indhold af følgebrevet.
Ved en eventuel re-submission skal det oplyses hvilke ændringer der er foretaget i den forbindelse.
Vi opfordrer til at CTCG’s Templates for følgebreve anvendes. Den seneste udgave, kan findes på CTCG’s hjemmeside under bjælken "Key documents list".
6. Skal jeg stadig sende min ansøgning til Lægemiddelstyrelsen og det Videnskabsetiske Komité System efter forordningen er trådt i kraft?
Den kliniske forsøgsansøgning behandles hos både os og det Videnskabsetiske Komité System ved indsendelse i den nye EU-portal. Derfor skal der ikke indsendes ansøgninger direkte til Lægemiddelstyrelsen eller det etiske komitésystem. Dette gælder dog kun for lægemiddelforsøg.
7. Hvilke dokumenter skal indsendes på dansk?
I Annex II til EU-Kommissionens spørgsmål og svar dokument fremgår hvilke dokumenter der kan indsendes på dansk eller engelsk for Part I. Spørgsmål og svar dokumentet findes under Chapter V - Additional documents i den opdaterede EudraLex Volume 10 til forordningen.
8. Skal de samme dokumenter uploades flere gange?
Fremadrettet kan det skabe problemer i CTIS portalens struktur, hvis det samme dokument er uploadet i flere sektioner. I stedet kan det aktuelle dokument uploades i én sektion og et blankt dokument med reference, uploades i de øvrige sektioner.
9. Skal forsøg der kun skal foregå i Danmark (danske mononationale forsøg, inklusive forsøg fra non-kommercielle sponsorer) indsendes gennem EU portalen?
Alle lægemiddelforsøg skal indsendes gennem EU portalen.
10. Skal alle indsendelser ske gennem portalen, fx årlige sikkerhedsrapporter?
Alle indsendelser i løbet af forsøgets levetid inklusive notifikationer, væsentlige ændringer, årlige sikkerhedsrapporter, afsluttende rapport og resultater skal ske gennem portalen. En undtagelse er dog SUSARS som skal sendes direkte til EudraVigilance databasen. EMA har udarbejdet vejledning og organiseret træning.
11. Er det muligt at henvise til kvalitetsdokumentation fra andre forsøg eller henvise til kvalitetsdokumentation der indsendes separat af fremstiller, for at sikre konfidentialitet?
Det er muligt at henvise direkte til et andet forsøg, der er godkendt under forordningen eller allerede overført til CTIS, hvor den samme kvalitetsdokumentation er vurderet. Alternativt er der aftalt en procedure, hvor fremstilleren kan indsende kvalitetsdokumentationen (IMPD-Q) separat i CTIS. Begge disse scenarier er beskrevet under spørgsmål 2.15 i EU Kommisionens spørgsmål/svar-dokument under Chapter V - Additional documents i den opdaterede EudraLex Volume 10 til forordningen.
12. Hvad skal jeg være opmærksom på, hvis mit forsøg er et genterapiforsøg?
Patientforsøg med præparater, der indeholder levende, genetisk modificerede organismer, er omfattet af "lov om miljø og genteknologi", specielt lovens regler om forskning. Reglerne fremgår af Arbejdsministeriets bekendtgørelse (nr. 910 af 11. september 2008) om "genteknologi og arbejdsmiljø".
Bekendtgørelsen omfatter bl.a. krav til anmeldelse om klassifikation af lokaler, hvori hele eller dele af forsøget skal foregå, samt anmeldelse af projekter. Anmeldelserne sker for at sikre både arbejdsmiljøet og det ydre miljø. Arbejdstilsynet giver herefter en godkendelse af både lokaler og forsøg.
Der er indgået en samarbejdsaftale mellem Arbejdstilsynet og Miljøstyrelsen om behandling af visse anmeldelser om klassifikation af lokaler og anmeldelse af forskningsprojekter. Dette har ført til følgende praksis:
Anmeldelser om klassifikation af lokaler til genterapi og anmeldelse af projekter om anvendelse af levende, genetisk modificerede mikroorganismer til genterapi indsendes til Arbejdstilsynet, der så sender en kopi af anmeldelserne til høring i Miljøstyrelsen.
Lægemiddelforsøg med genterapi skal jf. ovenstående anmeldes til Arbejdstilsynet. Dette kan ske parallelt med ansøgningen til Lægemiddelstyrelsen.
Nærmere oplysninger om anmeldelser kan indhentes ved kontakt til Arbejdstilsynet (eller tlf. 39 15 20 00).
Clinical Trial Information System (CTIS)
EU-portalen (CTIS)
1 a) Hvordan skal jeg ansøge i EU-portalen (CTIS)?
Vi vil henvise til træningsprogrammet fra EMA. Her gennemgås alle trin ved video og vejledninger. Endvidere har GCP-enheden udarbejdet en CTIS-vejledning og EMA har udarbejdet Sponsors Handbook.
1 b) Hvor skal sponsor registreres for at anvende EU-portalen (CTIS)?
Sponsor skal være registreret i EMAs system til registrering af organisationer (OMS=Organizational Management Service”), nærmere information herom kan findes på EMAs hjemmeside med information vedrørende træning og forberedelse til anvendelse af CTIS.
1 c) Aktivering af e-mail notifikationer i CTIS
EMA har i juni 2026 udrullet en CTIS-forbedring, der gør det muligt for brugere at tilmelde sig e-mail-notifikationer fra CTIS.
Notifikationerne om meddelelser og advarsler (notices and alerts) i CTIS, sendes direkte til ens egen e-mailadresse.
Der udsendes en pulje e-mail-notifikationer i døgnet, typisk i løbet af natten, som indeholder en samlet liste over de meddelelser og advarsler (notices and alerts), der er indkommet i CTIS det sidste døgn. Brugere er stadig forpligtede til, aktivt at overvåge notifikationer CTIS. Dette er nødvendigt for at sikre, at man samme dag bliver opmærksom på tidsfølsomme opgaver, som f.eks. indkommende Request for Information (RFI).
Som standard er CTIS-brugere afmeldt e-mail notifikationer. For at tilmelde sig skal man aktivere den respektive skydeknap (slider) under sin personlige CTIS-profil.
Når funktionen er aktiveret, gælder indstillingen ensartet for alle meddelelser og advarsler, der er knyttet til ens CTIS-rolle(r).
⚠️Disclaimer: Lægemiddelstyrelsen vil, så vidt muligt, altid forsøge at vejlede i CTIS' funktioner efter bedste evne, men da CTIS ejes, drives og supporteres af European Medicines Agency (EMA), forbeholder vi os retten til at henvise til EMA ServiceNow.
EMA har opdateret Sponsor Handbook med de ovenstående instruktioner om hvordan notifikationerne aktiveres.
1 d) Er der i Danmark krav om registrering af Legal Repræsentant i CTIS-portalen?
Hvis Sponsor er bosiddende i et land udenfor EU, er det krav i Lov om Kliniske Forsøg med lægemidler §21, at der registreres en Legal Repræsentant med bopæl i EU i CTIS. Det er ikke tilstrækkeligt at ”Contact point for union” er bosiddende i EU.
1 e) Registrering af forsøgslægemidler i CTIS jf. protokol
Hvorledes håndteres det, hvis der ikke er overensstemmelse mellem den kliniske forsøgsprotokol, der angiver forsøgslægemiddel på substansniveau, og de registrerede forsøgslægemidler i CTIS, der angiver specifikke producenters lægemiddelprodukter?
Som udgangspunkt vil det være de forsøgslægemidler, der er angivet i protokollen, der skal anvendes i forsøget. Hvis forsøgslægemidlet er angivet på produktniveau (specifikt produkt) i CTIS, og protokollen bruger generiske produkter (ATC- eller substansniveau), stemmer CTIS og protokollen således ikke overens. I dette tilfælde, vil det være protokollen der er dikterende for brug af forsøgslægemiddel i forsøget. Sponsor opfordres dog til, at CTIS opdateres, så der er overensstemmelse mellem CTIS og protokollen, f.eks. ved at angive forsøgslægemidler på ATC-, substans, eller produktniveau tilsvarende niveauet for forsøgslægemidlerne, der anvendes i protokollen.
1 f) Hvordan tilknytter jeg forsøgslægemidler og placebo i CTIS-portalen i forbindelse med ansøgning om et nyt klinisk forsøg? OBS: Siden er under udarbejdelse.
Alle lægemidler/substanser, der anvendes i forsøget, skal være registreret i EMAs Extended EudraVigilance Medicinal Product Dictionary (XEVMPD), hvorefter det kan tilknyttes forsøget i CTIS. Placebo-produkt kan dog også tastes direkte i CTIS. For de lægemidler, der er registreret i XEVMPD, skal man for udviklingsprodukter kende EU MP number og EU substance number, for at kunne lave tilknytningen til forsøget i CTIS. Markedsførte produkter kan fremsøges fra CTIS vha. flere søgekriterier.
1 g) Hvordan skal jeg håndtere dokumenter med signatur i CTIS?
Såfremt man indsender signerede dokumenter (fx QP deklaration), skal disse uploades i en version ”not for publication” i tillæg til den version, der uploades ”for publication”. Se vejledning fra ACT EU her.
1 h) Hvor kan jeg se sagsbehandlingstiderne for min ansøgning?
I CTIS findes der en "Timetable" under den enkelte ansøgning (for initial ansøgning og efterfølgende ændringsansøgninger (substantial modifications). Denne timetable er automatisk genereret, og tager højde for fri- og helligdage i de berørte medlemslande. Samtidig er det værd at bemærke, at nogle frister er dynamiske da de f.eks. afhænger af hvornår vurderingen hos myndighederne er færdig. I kan læse mere om tidsfrister for ansøgninger i EMAs vejledning til CTIS i Sponsors Handbook (3.1.1. IN: general timelines).
1 i) Hvor kan jeg se om mit forsøg er godkendt i CTIS og hvilke versioner af dokumentationen der er godkendt?
Lægemiddelstyrelsen udsender ikke godkendelsesbreve for kliniske forsøg, da alt information om godkendelse af forsøget fremgår af CTIS. De godkendte versioner af forsøgsdokumentationen fremgår af den endelige assessment rapport, i henholdsvis Introduction og Conclusion afsnittene. Derudover kan sponsor downloade de strukturelle data fra CTIS som PDF-fil, hvori man kan finde navn på produkter samt estimeret end-of-trial dato. Sponsors Handbook viser hvordan man kan downloade struktureret data. Herefter kan man så klippe siden med end-of-trial dato ud af PDF-filen, hvis man ikke ønsker at dele yderligere oplysninger.
1 j) Hvor kan jeg få hjælp til tekniske spørgsmål vedrørende CTIS?
Dedikeret CTIS Helpdesk for ikke-kommercielle sponsorer
”The Accelerating clinical trials in the EU (ACT EU)” initiativ har etableret en dedikeret helpdesk, som tilbyder forskellige tiltag som støtte for ikke-kommercielle sponsorer til at navigere i det regulatoriske landskab der omgiver kliniske forsøg i EU.
CTIS helpdesk tilbyder bl.a. skræddersyet teknisk assistance til CTIS funktioner. Man indsender spørgsmål ved at oprette en "ticket" ved CTIS Service Desk, hvor tilhørsforholdet som ikke-kommerciel sponsor angives i det obligatoriske felt "User affiliation"
Vedrørende regulatoriske spørgsmål kan Lægemiddelstyrelsens sektion for kliniske forsøg (Send en e-mail) altid kontaktes direkte, ligesom GCP-enhederne også er behjælpelige ved spørgsmål.
Link: CTIS Service Desk
2. Væsentlige og ikke væsentlige ændringer og underretninger (Subtantial- & Non-substantial Modifications)
2 a) Er det stadig muligt at indsende en ny væsentlig ændring, når jeg allerede har en ændring under sagsbehandling i CTIS?
For indsendelse af væsentlige ændringer i CTIS, anbefaler vi, at læse videre her: Substantial Modifications.
2 b) Hvilke krav gælder for underretning i CTIS portalen før, under og efter forsøget?
Medlemslandene skal notificeres om følgende i CTIS-portalen:
- Forsøgets start
- Start af rekruttering
- Afslutning af rekruttering
- Afslutning af forsøg (nationalt og globalt)
- Forventet dato for publicering af Summary of Results
- Tredjelandsinspektioner
Hvis der opstår sikkerhedsmæssige hændelser, fx Temporary halt, Urgent Safety Measures, Unexpected Events eller lignende, skal Sponsor underrette medlemslandene i CTIS.
Vejledning findes i Sponsors Handbook
2 c) Ikke væsentlige ændringer
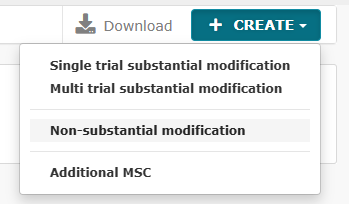
Det er nu blevet muligt at indsende ikke-væsentlige ændringer, der førhen skulle indsendes via en substantial modification.
Dette indbefatter blandt andet forlængelse af forsøgsperioden.
Se den fulde liste af nye CTIS funktioner for ikke-væsentlige ændringer længere nede på siden.
Der er to typer ikke-væsentlige ændringer:
- Non-substantielle ændringer (NSM)
- 81.9 non-substantielle (81.9 NSM)
81.9-NSM skal opdateres løbende af sponsor i CTIS i løbet af forsøgsperioden.
Andre NSM’er kan opdateres i CTIS sammen med den næste væsentlige ændring (SM).
81.9-NSM er ændringer i et klinisk forsøg, som ikke er væsentlige ændringer (SMs), men som ikke desto mindre er relevante for de berørte medlemsstaters tilsyn med de kliniske forsøg, skal løbende opdateres af sponsoren i EU-databasen i overensstemmelse med artikel 81, stk. 9, i forordning (EF) nr. 536/2014. For en liste over ikke-væsentlige ændringer henvises til bilag IV i dette dokument: CLINICAL TRIALS REGULATION (EU) NO 536/2014, VERSION 7.1
Det er altid sponsors ansvar at vurdere, om en ændring er non-substantiel (at den ikke påvirker forsøgets videnskabelige værdi eller deltagernes rettigheder og sikkerhed). Det er vigtigt, at denne vurdering kan begrundes ved monitoreringsbesøg og eventuelle inspektioner.
Alle ikke-væsentlige ændringer, både 81.9-NSM og NSM, kræver ikke vurdering og godkendelse af myndighederne før implementering.
Se den fulde liste af nye CTIS funktioner for ikke-væsentlige ændringer herunder:
Fanen MSCs:
- Trial subject numbers
Fanen Part I:
- Trial information - følgende sektioner er nu redigerbare;
- Medical condition
- Trial duration
- Source of monetary support or material support
- Age range (mindreårige kan ikke tilføjes)
- Clinical trial group (Healthy, patients, vulnerable)
- Protocol
- Alle sektioner er redigerbare, nye dokumenter kan uploades, nuværende dokumenter kan opdateres
- Alle sektioner er redigerbare, nye dokumenter kan uploades, nuværende dokumenter kan opdateres
- SA and PIP
- SA kan tilføjes/opdateres
- PIP kan opdateres, hvis der er en eksisterende, men der kan ikke tilføjes en ny
- Associated CTs
- Associated CTs kan tilføjes/slettes
- Document agreement from another sponsor, kan tilføjes/opdateres
- Sponsor
- Sponsorinformationer kan ændres, så længe det ikke er ORG-ID
- Co-sponsor kan slettes, men ikke tilføjes
- Responsibilities kan ændres
- Contact points kan tilføjes/ændres
3. Transparens
Hvor meget bliver offentliggjort i EU-portalen og databasen (CTIS)?
I overensstemmelse med de reviderede CTIS transparensregler (Revised CTIS transparency rules (europa.eu), er det kun visse dokumenter, som uploades i EU-databasen, der offentliggøres. Det har blandt andet til formål at beskytte personoplysninger og kommercielt fortrolige oplysninger.
Pr. 18. Juni 2024 er CTIS tilpasset de reviderede CTIS transparensregler.
Vi henviser derfor til ECT EU Guidance. Endvidere til quick guide for users, der giver et hurtigt overblik over hvilke dokumenter, der bliver offentliggjort hvornår.
Generelt skal al inklusion af persondata minimeres og såfremt der inkluderes persondata i dokumentversion ”for publication”, skal disse anonymiseres med få undtagelser, hvor informationen er påkrævet at blive publiceret.
4. Re-submission
Hvordan indsendes en re-submission i CTIS?
En eventuel re-submission ansøges i CTIS ved hjælp af ”re-submission”-funktionaliteten på det eksisterende EU CT forsøgsnummer. Det skal fremgå af cover letter at der er tale om en re-submission.
Vi anbefaler at man, i alle uploadede dokumenter indeholdende EU CT nummer, fjerner de to sidste cifre i nummeret, da disse ændre sig ved hver re-submission. På denne måde undgår man at skulle opdatere dette nummer igen, ved evt. ny re-submission.
Man kan med fordel, ved re-submission, lave de rettelser der blev stillet considerations om ved initial indsendelse af forsøget. Man skal blot huske at adressere disse ændringer i cover letter, og tydeliggøre hvor i dokumentationen opdateringerne findes. Herudover skal opdaterede dokumenter uploades i både en clean version og track changes version.
For vejledning til resubmission se CTIS Sponsor Handbook - 2.72. Resubmitting an INog Rapportering af SUSARs og årlige sikkerhedsrapporter
For rapportering af sikkerhedsmæssige indberetninger anbefaler vi at læse videre her: ”Sikkerhedsrapportering”
GCP-afvigelser
GCP-afvigelser
Hvilke krav gælder for rapportering af GCP-afvigelse (serious breaches)?
Jf. EU-forordningen vil der også være krav om at rapportere alvorlige GCP-afvigelser (læs definition på vores side vedr. alvorlige afvigelser). Fremover skal alvorlig afvigelse fra protokol/forordning rapporteres i EU-portalen, I kan se mere i modul 5 under Sponsor Workspace i EMAs online træning.
Krav til prævention og graviditetstestning i kliniske forsøg med lægemidler
Krav til prævention i kliniske forsøg
Kravene til brug af prævention i kliniske forsøg adskiller sig fra dem i almindelig klinisk praksis. I kliniske forsøg har sponsor en forpligtigelse til at minimere risikoen for eksponering af et potentielt foster. I kliniske forsøg er der derfor særlige krav til graviditetstest og brug af prævention for at sikre patienters sikkerhed og undgå unødig risiko for et potentielt foster
Det er derfor essentielt, at forsøgsprotokollen indeholder en detaljeret beskrivelse og begrundelse for, hvordan præventionskrav og graviditetstestning håndteres i det specifikke kliniske forsøg.
Generel reference bør ske til Clinical trials Coordination Group (CTCG) vejledning som detaljeret gennemgår de fælles EU anbefalinger til præventionsmetoder og graviditetstestning i kliniske forsøg.
Vejledningsdokumentet fra CTCG Clinical Trials Coordination Group "Recommendations related to contraception and pregnancy testing in clinical trials Version 1.2" findes på HMAs hjemmeside under Key Documents list og afsnittet Other Guidance and Q&A Documents.
Krav ved inklusion af mænd i forsøg
Seksuelt aktive mænd med fertil kvindelig partner skal som minimum anvende kondom så længe, at det ikke kan afvises, at forsøgslægemidlet kan udgøre en risiko for et foster. Hvis der i Investigator’s Brochure (IB) eller produktinformationen for et godkendt lægemiddel er beskrevet påvist embryoføtal toksicitet, skal den fertile kvindelige partner til den mandlige forsøgsdeltager opfordres til at bruge sikker prævention i kombination med kondom.
Krav ved inklusion af fertile kvinder i forsøg
Det skal fremgå tydeligt af protokollen, eller tillæg hertil, at fertile kvinder testes for graviditet inden deltagelse (negativ graviditetstest påkrævet). Fertile seksuelt aktive kvinder skal anvende sikker prævention (highly effective methods of contraception) ved deltagelse i kliniske forsøg, indtil der er indsamlet nok data til at foretage en tilfredsstillende analyse af de potentielle risici, som et forsøgslægemiddel under udvikling udgør for et foster og for forsøgslægemiddel med identificeret høj risikoprofil (påvist reproduktions/genotoksicitet og/eller teratogenicitet).
Følgende metoder anses som sikker prævention: spiral (med eller uden hormon), hormonel antikonception (p-piller, implantat, transdermale depotplastre, vaginalring eller depotinjektion) eller abstinens (kun acceptabelt hvis det er i overensstemmelse med forsøgspersonens normale livsførelse). I særlige tilfælde kan andre præventionsmetoder accepteres, hvilket forudsætter en grundig faglig begrundelse med udgangspunkt i forsøgsdesign, lægemidlets toksicitetsprofil, påvirkning på hormonel prævention og/eller patientpopulation.
Såfremt der er tilstrækkelig dokumentation for, at lægemidlet ikke er reproduktionstoksisk i mennesker, kan mindre sikre præventionsmetoder end de tidligere nævnte være tilstrækkelige. Dette er nærmere beskrevet i CTCGs vejledning som findes på HMAs hjemmeside under Key Documents list under afsnittet Other Guidance and Q&A Documents.
Præventionskrav under og efter forsøget
Fertile kvinder og mænd, der inkluderes i forsøg, skal anvende midler til prævention i overensstemmelse med CTCGs vejledning i hele forsøgsperioden og efter forsøgsophør i en periode, indtil den systemiske eksponering af forsøgsmedicinen er faldet til en koncentration, der ikke er relevant for human teratogenicitet. Som udgangspunkt defineres denne periode som fem gange lægemidlets halveringstid efter sidste dosis eller det tidsinterval, der evt. er beskrevet i forsøgslægemidlets produktinformation eller Investigator’s Brochure.
Protokollen skal tydeligt omfatte:
- En korrekt definition af fertile kvinder / women of childbearing potential (WOCBP)
- Beskrivelse af forsøgslægemidlets toksicitetsprofil (reproduktion, genotoksicitet, teratogenicitet )
- Begrundelse for valg af påkrævede præventionsmetoder baseret på forsøgslægemidlets toksicitetsprofil samt en udspecificeret liste over acceptable præventionsmetoder
- Overvejelser om interaktioner, der påvirker hormonbaseret prævention
- Angivelse af varighed af brug af prævention (typisk indtil fem gange halveringstiden af lægemidlet efter sidste dosis)
- Som minimum graviditetstest ved inklusion, graviditetstest er som oftest også et krav under forsøget indtil ophør af klinisk relevant eksponering.
- Tidspunkter og frekvens for yderligere graviditetstest baseret på toksicitetsprofilen for forsøgslægemidlet
- Relevante in- og eksklusionskriterier for præventionskrav, graviditet og amning
- For mandlige forsøgsdeltagere stillingtagen til behov for brug af kondom og/eller krav om brug af prævention for WOCBP-partner
Link til skabelon til protokollen til finder du her.
Deltagerinformation
Deltagerinformationen skal altid afspejle protokollens præventionskrav inkl. hvilke præventionsformer, der anses for acceptable i forsøget og graviditetstestning samt formidle information om potentielle toksicitetsrisici. Hvis relevant skal forsøgsdeltagere informeres om forbud mod sædcelle/æg donation og varighed af dette forbud. Der skal også overvejes, om der er behov for information om og procedure for nedfrysning af sæd/æg for fremtiden.
Undtagelser
Sterile eller ikke fertile forsøgsdeltagere er undtaget fra præventionskrav. Sterilitet defineres normalt ud fra kirurgisk steriliseret (vasektomi/bilateral tubektomi, hysterektomi og bilateral ovariektomi) eller postmenopause defineret som udebleven menstruation i mindst 12 måneder uden anden årsag og bekræftet ved måling af FSH-niveauet.
Særlige forhold
Der kan for enkelte individer eller særlige populationer være forhold, som taler imod brug af ovenstående metoder. Eksempler kan være alvorligt svækkede hospitaliserede patienter eller ikke seksuelt aktive fertile børn. Der skal i protokollen være en robust retfærdiggørelse for eventuelle undtagelser og afvigelser fra CTCGs vejledning med afsæt i, at det er nødvendigt for, at forsøget kan gennemføres.
Andre emner
Kan man deltage i flere kliniske forsøg på samme tid?
Det er den kliniske forsøgsprotokol der skal tage stilling til, hvorvidt inklusion i forsøget kan tillades ved samtidig behandling i et andet klinisk forsøg.
I nogle protokoller er der inkluderet et standardeksklusionskriterie, som udelukker inklusion af individer der allerede deltager et andet klinisk forsøg. Nye typer af forsøgsdesigns, fx lav-interventions clusterforsøg (hvor forsøgslægemidlerne har en markedsføringstilladelse, såsom influenzavaccination), kan udfordre denne type af generel eksklusion, da de nye lav-interventions forsøgsdesign kan inkludere meget store populationer, som derved sætter en væsentlig begrænsning for rekruttering. Det bør derfor overvejes, at eksklusionskriteriet for deltagelse i andre forsøg er fleksibelt og vurderes ad hoc.
Det er vores vurdering, at der er tale om en non-substantiel ændring hvis en forsøgsprotokol opdateres med et fleksibelt eksklusionskriterie for at imødekomme ovenstående udfordring.
Hvad skal jeg gøre, hvis jeg ønsker, at DK påtager sig rollen som RMS?
Krav til indsendelse
Det er vores generelle tilgang, at vi gerne vil påtage os opgaven som RMS, når en sponsor foreslår os i CTIS.
Man behøver således ikke at sende mail på forhånd, da vi vil gøre vores bedste for at imødekomme ønsket. Der kan dog være perioder hvor vi er nødsaget til at afslå, f.eks. såfremt vi har mange forsøg, der modtages i samme periode.
Hvordan skal jeg indsende min ansøgning efter forordningen er trådt i kraft?
Under EU forordningen om kliniske forsøg med humanmedicinske lægemidler, skal alle forsøg ansøges i et fælles europæisk system (CTIS). Det betyder, at al information om forsøg i EU er samlet ét sted. Ved forsøg, der ønskes gennemført i flere lande, samarbejder de berørte landes myndigheder om vurderingen. På nationalt plan koordinerer Lægemiddelstyrelsen og etiske komiteer en samlet afgørelse.
Hvor kan jeg læse mere om Scientific Advice?
Læs mere på Lægemiddelstyrelsens side: Videnskabelig rådgivning (scientific advice) om lægemidler
Hvis jeg planlægger et komplekst forsøg, hvor kan jeg finde vejledning?
CTCG har samlet Anbefalinger og Spørgsmål/svar på deres hjemmeside. De kan findes under Key document list.
Et direkte link til recommendation paper on complex clinical trials findes her.
Spørgsmål/svar vedrørende kliniske lægemiddelforsøg under Direktivet
Spørgsmål/svar i forbindelse med ansøgning om kliniske lægemiddelforsøg under direktivet
1. Hvordan skal jeg rapportere mine resultater til Lægemiddelstyrelsen?
For forsøg godkendt under direktivet, skal forsøgsresultater indtastes i EudraCT snarest muligt og senest inden 1 år efter forsøgets afslutning. Data vil herefter blive offentliggjort på EU Clinical Trials Register. Dette erstatter, at forsøgets resultater skal indsendes til os. Man kan læse mere på EudraCT hjemmesiden under ’update’ og generelt om afslutning af forsøg i vores vejledning afsnit 13. De offentlige GCP-enheder har lavet en vejledning, som kan findes her.
Forsøg godkendt under direktivet og som ikke overføres til klinisk forsøgsforordningen er der stadig krav om at resultaterne for disse forsøg skal registreres i EudraCT. EudraCT vil stadig være tilgængelig for upload af resultater.
2. Hvordan fremsøger jeg de forsøg, hvor der ikke er offentliggjort resultater i EudraCT/clinicaltrialsregister.eu?
Man kan søge på EU Clinical Trial Register på fx det relevante sygehus. Her fremgår det hvilke forsøg (Trial protocol, DK) der er registreret som afsluttede (completed) og hvis der mangler resultater, er der anført Trial results: No results available
(Man skal dog være opmærksom på at afdeling mv. skal være stavet korrekt i det oprindelige EudraCT skema for at kunne fremsøges. Det kan derfor være en fordel, ikke at søge for specifikt).
Ændringslog:
30. juni, 2026: CTIS punkt opdateret med aktivering af e-mail notifikationer fra CTIS
21. maj, 2026: CTIS punkt opdateret med "Registrering af forsøgslægemidler i CTIS jf. protokol (punkt 1.d)
22. april, 2026 - Link til EMA FAQ for clinical trial sponsors under Requirements for documents, afsnit 3
9. februar, 2026 - Info om ny mulighed for at håndtere ikke væsentlige ændringer i CTIS
4. november, 2025 - Præcisering af sponsordefinition
14 oktober, 2025 - Revideret protokolskabelon, der reflekterer ICH E6 R3 GCP standard og sponsor oversight
6. oktober, 2025 - Tilføjelse af Q&A om Sponsoransvar
14. juli, 2025 - Opdatering til ny CTIS Sponsorhåndbog v. 6.
2. april, 2025 - Definitioner på lav-interventionsforsøg og pragmatiske elementer tilføjet.