
Hvad er et klinisk forsøg?
Kliniske forsøg er afprøvninger af medicins (lægemidlers) virkning og bivirkninger
Et klinisk forsøg gennemføres for at finde ud af, hvordan et lægemiddel virker, hvilke bivirkninger det har, og hvordan det omsættes i kroppen.
Forsøg med lægemidler foregår i forskellige faser, som er en overordnet inddeling af de forskellige udviklingstrin. Fase I, II og III er typisk forsøg med ikke-godkendte lægemidler under udvikling. Fase I og II forsøg inkluderer i udgangspunktet relativt få forsøgspersoner, hvor fase III potentielt kan inkludere tusindvis. Fase IV er forsøg med allerede godkendte lægemidler, der undersøger den eksisterende brug af lægemidlet eller sammenlignelige behandlinger. Læs mere her: ICH E8 General considerations for clinical studies - Scientific guideline | European Medicines Agency (europa.eu)
Grundlæggende arbejder Lægemiddelstyrelsens for at deltagerne i kliniske forsøg med lægemidler, kan være sikre på at der er styr på sikkerheden og at forsøget kan give gode robuste resultater.
Detaljeret beskrivelse af definitioner på kliniske forsøg kan du finde som punkt under vores Spørgsmål & Svar fane.
Frivillige forsøgspersoner afprøver lægemidler
Kliniske forsøg kan inkludere enten raske, frivillige forsøgspersoner eller frivillige patienter afhængigt af hvad der undersøges. Alle forsøgspersoner i kliniske forsøg skal have både mundtlig og skriftlig information om forsøget og give deres skriftlige samtykke til at deltage, før de starter i forsøget. I helt særlige tilfælde kan der benyttes andre former for samtykkeformer, hvilket der kan læses mere om hos de Videnskabsetiske Medicinske Komitéer (VMK).
Kliniske forsøg skal godkendes
I Danmark skal kliniske forsøg godkendes af Lægemiddelstyrelsen og de Videnskabsetiske Medicinske Komiteer før det må påbegyndes. Den overordnede ansvarlige for forsøget (sponsor) laver en ansøgning og sender den ind til EU-portalen Clinical Trial Information System (CTIS). Når ansøgningen sendes ind i CTIS sendes den til både Lægemiddelstyrelsen og VMK. Lægemiddelstyrelsen foretager en grundig vurdering af forsøgets kvalitet og sikkerheden for forsøgspersonerne. VMK vurderer blandt andet forsøgets etiske aspekter og det videnskabelige grundlag.
Lægemiddelstyrelsens sektion for Kliniske Forsøg (KF) koordinerer en fælles vurdering af part I i CTIS. Part II vurderes udelukkende af de Videnskabsetiske Medicinske Komiteer(VMK).
I Danmark er VMK centraliseret ved Nationalt Center for Etik, som koordinerer den videnskabsetiske vurdering af kliniske forsøg. Læs mere på VMKs hjemmeside.
Det effektive samarbejde internt mellem VMK og KF giver ansøgere let adgang til hurtig og kompetent hjælp både før, under og efter godkendelsesprocessen.
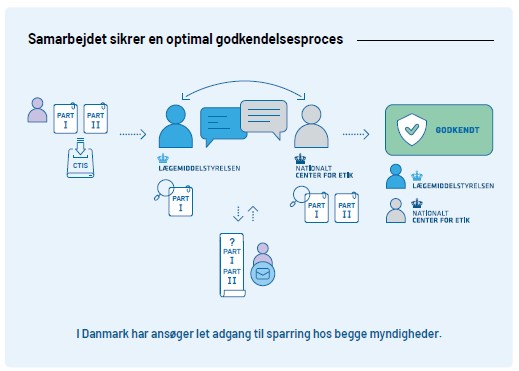
Se her for en højere opløsning af illustrationen af godkendelsesprocessen.
Før et klinisk forsøg kan starte, skal både VMK og Lægemiddelstyrelsen have godkendt forsøget.
Den centrale database for kliniske forsøg – Clinical trial Information System (CTIS)
CTIS er administreret af European Medicines Agency (EMA); den centrale Europæiske Lægemiddelstyrelse, men det er de nationale medlemsstaters lægemiddelstyrelser og etiske komiteer der fælles laver vurderingen af de kliniske forsøg.
CTIS er det centrale sted for indsendelse og de efterfølgende korrespondancer mellem myndighederne og sponsor vedrørende de spørgsmål myndighederne måtte have før forsøget kan godkendes og til ansøgers (sponsors) svar. Afgørelsen lægges ligeledes i CTIS.
For forsøg der parallelt ansøges i flere lande (multinationalt) koordineres godkendelsesprocessen af en af medlemsstaterne som er valgt som Rapporterende Member State (RMS). Denne RMS koordinerer den samlede vurdering, som præsenteres i CTIS.
Det er muligt for alle at søge information om forsøg i Danmark og EU i CTIS-databasen. Der kan læses mere om hvornår hvilken information om kliniske forsøg offentliggøres, her.
Forsøg der er godkendt før 31. januar 2022 skal søges i registeret for det daværende system (clinicaltrialsregister).
Inspektion af kliniske forsøg
Lægemiddelstyrelsen udfører inspektioner af kliniske forsøg for at sikre, at udførslen overholder den givne godkendelse samt retningslinjerne for god klinisk praksis (GCP).
Lægemiddelstyrelsen foretager inspektioner af kliniske forsøg med lægemidler i både Danmark og i udlandet, herunder inspektioner koordineret via Det Europæiske Lægemiddelagentur (EMA).
Du kan læse mere om GCP-inspektioner her.
Bivirkninger i kliniske forsøg med lægemidler
De forsøgsansvarlige læger (investigator) skal indsamle og registrere bivirkninger i henhold til forsøgets protokol. Hvis der opstår alvorlige uventede bivirkninger skal sponsor straks rapportere til EudraVigilance (EU's bivirkningsdatabase). Lægemiddelstyrelsen overvåger disse i EudraVigilance. Du kan læse mere om bivirkningshåndtering i kliniske forsøg og hvordan disse rapporteres her.
Samarbejde i EU om kliniske forsøg
De Europæiske Lægemiddelstyrelser er organiseret i Heads of Medicines Agencies (HMA) og herunder er Clinical Trial Coordination Group (CTCG) en gruppe hvor Danmark er meget aktiv i arbejdet for at harmonisere og styrke godkendelsesproceduren mellem medlemsstaterne og udarbejde vejledning til ansøgerne (sponsorerne).
Der foregår også tværeuropæisk samarbejde om at fremme og optimere kliniske forsøg under den Europæiske lægemiddelstyrelse EMA , selvom godkendelsen af kliniske forsøg er et nationalt anliggende. Se mere om programmet Accelerating clinical trials in the EU her.
Spørgsmål og svar
Vi har udarbejdet nogle spørgsmål og svar vedr. kliniske forsøg med lægemidler, som blandt andet forklarer definitioner på kliniske forsøg og besvarer hvorvidt et konkret forsøg skal ansøges til Lægemiddelstyrelsen, som du kan finde her: Spørgsmål og svar
Hvis du ikke finder svar her kan du altid ringe til os eller sende en mail.